第三节 肝胆疾病
一、循环障碍
(一)门静脉阻塞
较为少见。多由于肝、胰疾病如肝硬变、肝癌、胰腺癌等压迫、侵袭肝内门静脉,以及化脓性腹膜炎,新生儿脐带感染化脓等引起门静脉的血栓形成或栓塞。门静脉完全而广泛的阻塞甚少见。其肝内分支的一支或多支阻塞可引起梗死(Zahn梗死)。
Zahn梗死 又称萎缩性红色梗死,为肝内少见的循环障碍性病变。病变以局部肝淤血为主,而不是真性梗死。病变区呈圆形或长方形,暗红色,境界清楚(图10-32)。镜下为肝小叶中央区的高度淤血并有出血。局部肝细胞萎缩、坏死或消失。本病变对机体无大影响,偶可成为腹腔内出血的来源。病变恢复期可见阻塞的门静脉周围出现新吻合支。

图10-32 肝Zahn梗死
梗死区略呈长方形,色深,被膜下肝组织未发生梗死
(二)肝静脉阻塞
肝静脉阻塞一般分为二类。一类为肝静脉干至下腔静脉的阻塞,称Budd-Chiari综合征;另一类为肝内肝静脉小分支阻塞,称肝小静脉闭塞症(veno-occlusive disease)。
Budd-Chiari综合征 是指肝静脉干和(或)下腔静脉的肝静脉入口处有一段完全或不完全阻塞而引起的征候群。本综合征的病因有原发及继发二种。原发性者主要是先天性血管异常,如下腔静脉膜性阻塞所致的肝静脉阻塞。继发性者可由血液凝固性积升高疾病(如红细胞增多症),肝癌及腹腔肿瘤,腹部创伤及某些口服避孕药等引起的该段静脉血栓形成所致。病理变化主要为肝淤血,肝细胞萎缩、变性以至坏死。此外,还有肝出血,即淤于肝窦内的红细胞进入窦外压力较低的Disse腔及萎缩的肝板内。慢性病例则发展为淤血性肝硬变。
二、代谢性疾病
(一)含铁血黄素沉积症
肝含铁血黄素沉积症(hemosiderosis)是指肝组织内有可染性铁的血色素沉着。含铁血黄素沉积的病因,主要是由于大量红细胞破坏,血红蛋白分解所引起,如引起溶血及肝内出血的疾病(慢性溶血性贫血)。含铁血黄素主要沉积于肝细胞内,Kupffer细胞内亦常有该色素沉着但一般较肝细胞轻。因输血引起者Kupffer细胞色素沉着则较明显。
血色病(hemchromatosis)是一种先天性铁代谢异常的全身性疾病。发病机制不明。肝病变为全身病变的一部分,表现为肝内重度含铁血黄素沉积,全肝呈铁锈色。后期伴有肝纤维化或肝硬变。
(二)糖原沉积症
糖原沉积症(glycogenosis)为先天性常染色体隐性遗传所引起的组织内糖原质的异常和量的增多,而引起沉积。主要累及肝、心、肾及肌组织,有低血糖、酮尿及发育迟缓等表现。按糖代谢过程中不同环节、不同酶的异常,现已将本症分为O~Ⅺ型,如包括亚型则更多(Schiff,1987)。
肉眼可见肝肿大,有的可达正常肝的3倍以上。颜色变淡。镜下,肝细胞明显肿大,泡浆淡染,呈疏松的颗粒状并有空亮区。冷冻切片,PAS染色可见肝细胞内红色的糖原颗粒。对淀粉酶的消化反应稳定。后期,多种类型可伴有肝纤维化或肝硬变。需要指出,确诊糖原沉积症及分型,不能单凭病理组织学改变,必须结合临床及用肝穿取新鲜标本作酶类分析。
(三)类脂质沉积症
类脂质沉积症(lipoidosis)是先天缺陷性脂质代谢障碍所致的组织内类脂质增多并沉积。主要有糖脂、磷脂及胆固醇等沉积。其发生机制,大都是由于作用于脂质分解代谢某些环节上的酶类的遗传性缺失,使其相应的底物(脂质)分解代谢不能进行而沉积在组织内。
1.糖脂沉积症 糖脂是指不含磷酸的脑苷脂及神经苷脂等脂类。它们的分解代谢障碍可分别引起脑苷脂沉积症(如高雪病)和神经节苷脂沉积症。
高雪(Gaucher)病,也称脑苷脂沉积症,是由于常染色体隐性遗传所致体内β-葡萄糖苷酶缺乏而引起的脑苷脂分解代谢障碍。主要累及肝、脾、淋巴结及骨髓等单核吞噬细胞系统。常发生在婴儿,为致命性疾病。主要病变为肝、脾肿大,脾大尤为明显,可达正常脾重的20倍。镜下,肝内聚集大量高度胀大的载脂巨噬细胞,有的胞浆呈泡沫状,有的胞浆出现红染条纹,后者排列成皱纹纸样外观,胞核小,圆形或椭圆形居于细胞中央,称为高雪细胞(图10-33)。这些细胞主要分布于小叶中央静脉附近的肝窦内和汇管区。偶见发生肝纤维化和肝硬化。

图10-33 高雪病之肝
图中央区为数个高雪细胞,位于肝窦内胞浆呈皱纹纸状外观 ×400
2.磷脂沉积症 主要为不含甘油成分的神经磷脂的增多、蓄积,又称尼曼-皮克病。
尼曼-皮克(Niemann-Pick)病又称神经磷脂沉积症。系由于常染色体隐性遗传所致的神经磷脂酶缺乏,使神经磷脂不能被水解而沉积于组织内所致。另外还可伴有其它脂质贮积。本病主要累及肝、脾、骨髓及淋巴结等器官,在儿童也侵犯神经系统。肝病变肉眼观,肝肿大。镜下,在肝窦内和汇管区有大量Kupffer细胞和巨噬细胞聚集,细胞体积肿大,胞浆呈泡沫状,核小居中,称为Pick细胞。肝细胞内也可见有脂肪,主要为中性脂肪及胆固醇。电镜下见Pick细胞内充满多数年轮样层状排列的球形包涵体。本病常发生于幼儿,预后不佳。
(四)铜代谢障碍疾病―肝豆状核变性
肝豆状核变性(hepatolenticular degeneration)又称威尔逊病(Wilson’s disease)。本病为位于第13染色体的隐性基因传递的遗传性疾病,家族性多发。患者多为儿童及青少年。本病的特点是铜代谢障碍,不能正常排出而蓄积于各器官。首先累及肝,待肝饱和后再沉积于中枢神经系统,故出现神经症状。铜也可蓄积于角膜,在角膜周围出现绿褐色环(Kayser-Fleischer环)。肝病变:在肝细胞中可见有脂褐素、铜结合蛋白、铁等沉着。铜或铜结合蛋白(如rhodamine,rubeanic acid等)可由组织化学染色检出。早期见肝细胞线粒体基质中有大颗粒或晶体沉着。肝并有急、慢性肝炎及肝硬变等病变伴发。中枢神经系统可见被壳及苍白球变性,尾状核萎缩,大脑皮质及小脑变性等。
三、病毒性肝炎
病毒性肝炎(viral hepatitis)是由肝炎病毒引起的以肝实质细胞变性坏死为主要病变的传染病。现已知肝炎有甲型、乙型、丙型、丁型及戊型5种,由各该型病毒引起。1974年以来提出的非甲非乙型肝炎(NANB型),经近年研究证明,其中大部分为丙型肝炎并检出了丙肝病毒(HCV)及其抗体,另一小部分则为戊型肝炎。肝炎在世界各地均有发病和流行,且发病率有不断升高趋势。其发病无性别差异,各种年龄均可罹患。
病因
从1970年起,经过20年的研究,目前对肝炎病毒已比较清楚,由最初仅知的甲型肝炎病毒和乙型肝炎病毒二种,增加到由甲到戊5种病毒(HAV~HEV)。其特点见表10-2。
表10-2 各型肝炎病毒特点
| 肝炎及其病毒分型 |
病毒类型 |
传染方式 |
克隆化年代 |
|
甲型肝炎
HAV |
Enterovirus
RNA型 |
肠道 |
1983年(1973检出) |
|
乙型肝炎
HBV |
Hepadnavirus
DNA型 |
非肠道 |
1979年(1963检出) |
|
丙型肝炎
HCV |
Flavivirus
RNA型 |
非肠道 |
1989年 |
|
丁型肝炎
HDV |
Viroidrelated
RNA型 |
非肠道 |
1986年 |
|
戊型肝炎
HEV |
Calcivirus
RNA型 |
肠道 |
1990年 |
(采自Seeff)<
在上述各型肝炎病毒中,以HBV发现最早,研究得最多。现知该病毒是由核心及外壳两部分构成的病毒颗粒(Dane颗粒)。病毒基因组DNA在肝细胞核内进行复制、转录,合成核心颗粒后,被转运到肝细胞浆内,在通过内质网和细胞膜时合成其外壳部分,并以发芽过程释出肝细胞。Dane颗粒的核心部分含核心抗原(HbcAg),外壳部分含表面抗原(HBsAg)。目前认为引起肝细胞免疫损害的只是HBsAg诱发的免疫反应,其中以细胞免疫反应起主要作用。HAV是一种微小的RNA病毒,在肝细胞浆内增殖,现已被列入肠道病毒,该病毒颗粒可在粪便中检出。HCV的整组基因近年也可克隆化并标记其病毒抗体。HDV是一种微小缺陷性RNA病毒,现知此种病毒为球形以HBsAg作为其外壳,只能在HBsAg阳性机体内生长,故丁型肝炎常与乙型肝炎合并存在。HEV的颗粒为球形无包膜,表面有尖钉状突起。我国学者已将HEV分离培养并建立了细胞系(Huang RT等,1992)。
传染途径
各型肝炎病毒均可存在于肝组织、血液、粪、尿及各种体液内。其传染方式主要是经口、经血及体液传播,但各型肝炎的传染途径各异。甲型、戊型多经口感染。常来源于饮水及食物的污染,有时呈流行性暴发。乙型、丙型经血感染,主要通过输血、输液也可通过经皮及性接触传播。丁型亦为非经口感染,常与乙型肝炎传播伴行。各型肝炎的潜伏期也不相同,如甲型15~50天,乙型60~180天。戊型2~9周。丙型7~8周。一般认为肝炎痊愈后均可获得免疫力但均不稳固(甲型者稍好),有少部分患者还可发生再感染。
基本病变
各型肝炎病变基本相同,都是以肝细胞的变性、坏死为主,同时伴有不同程度的炎性细胞浸润、肝细胞再生和纤维组织增生。
1.肝细胞变性、坏死
(1)胞浆疏松化和气球样变:为常见的变性病变,是由于肝细胞受损后细胞水分增多造成。开始时肝细胞肿大,胞浆疏松呈网状、半透明,称胞浆疏松化。进一步发展,肝细胞更形胀大呈球形,胞浆几完全透明,称为气球样变(ballooning degeneration)(图10-34)。电镜下,可见内质网扩张、囊泡变、核蛋白颗粒脱失;线粒体肿胀、嵴消失等。

图10-34 急性病毒性肝炎
肝细胞胞浆疏松化和气球样变,肝窦受压变窄
(2)嗜酸性变及嗜酸性坏死:嗜酸性变多累及单个或几个肝细胞,散在于小叶内。肝细胞胞浆水分脱失浓缩,嗜酸性染色增强,胞浆颗粒性消失。如进一步发展,胞浆更加浓缩之外,胞核也浓缩以至消失。最后剩下深红色均一浓染的圆形小体,即所谓嗜酸性小体(acidophilic body或Councillman body)(图10-35)。上述改变称嗜酸性坏死(acidophilic necrosis),为单个细胞坏死(细胞凋谢)。

图10-35 急性病毒性肝炎
游离于肝窦内的嗜酸性小体(↑处),窦内皮细胞肿胀增生 ×800
(3)点状坏死(spotty necrosis):肝小叶内散在的灶状肝细胞坏死。每个坏死灶仅累及1至几个肝细胞。 同时该处伴以炎性细胞浸润(图10-36)。

图10-36 病毒性肝炎
肝细胞点状坏死,坏死灶内有炎性细胞浸润
(4)溶解坏死(lytic necrosis) 最多见,常由高度气球样变发展而来。此时胞核固缩、溶解、消失,最后细胞解体。重型肝炎时肝细胞的变性往往不明显,很快就发生此种坏死崩解。
2.炎细胞浸润 肝炎时在汇管区或肝小叶内常有程度不等的炎性细胞浸润。浸润的炎细胞主要是淋巴细胞、单核细胞,有时也见少量浆细胞及中性粒细胞等。
3.间质反应性增生及肝细胞再生
(1)Kupffer细胞增生肥大:这是肝内单核吞噬细胞系统的炎性反应。增生的细胞呈梭形或多角形,胞浆丰富,突出于窦壁或自壁上脱入窦内成为游走的吞噬细胞。
(2)间叶细胞及纤维母细胞的增生:间叶细胞具有多向分化的潜能,存在于肝间质内,肝炎时可分化为组织细胞参与炎性细胞浸润。在反复发生严重坏死病例,由于大量纤维组织增生可发展成肝纤维化及肝硬变。
(3)肝细胞再生:肝细胞坏死时,邻近的肝细胞可通过直接或间接分裂而再生修复。在肝炎恢复期或慢性阶段则更为明显。再生的肝细胞体积较大,核大而染色较深,有的可有双核。慢性病例在汇管区尚可见细小胆管的增生。
上述肝炎基本病变中,肝细胞疏松化,气球样变,点状坏死及嗜酸性小体形成对于诊断普通型肝炎具有相对的特征性;而肝细胞的大片坏死、崩解则是重型肝炎的主要病变特征。
临床病理类型
各型肝炎病毒引起的肝炎其临床表现和病理变化基本相同。现在常用的分类是,在甲、乙、丙、丁、戊5型病毒病因分类之外,把病毒性肝炎从临床病理角度分为普通型及重型二大类。在普通型中分为急性及慢性两类。急性有急性无黄疸型及黄疸型;慢性有持续性(迁延性)及活动性。重型中又分为急性及亚急性两种。
1.急性(普通型)肝炎 最常见。临床上又分为黄疸型和无黄疸型二种。我国以无黄疸型肝炎居多,其中多为乙型肝炎,一部分为丙型即过去所谓非甲非乙型的一部分。黄疸型肝炎的病变略重,病程较短,多见于甲型、丁型、戊型肝炎。两者病变基本相同,故一并叙述。
病变:广泛的肝细胞变性,以胞浆疏松化和气球样变最为普遍。坏死轻微,肝小叶内可有散在的点状坏死。嗜酸性小体的出现并非经常。由于点状坏死灶内的肝细胞索网状纤维支架保持完整而不塌陷,所以该处通过再生的肝细胞可完全恢复原来的结构和功能。汇管区及肝小叶内也有轻度的炎性细胞浸润。黄疸型者坏死灶稍多、稍重,毛细胆管管腔中有胆栓形成。
临床病理联系
由于肝细胞弥漫地变性肿胀,使肝体积增大,被膜紧张,为临床上肝大、肝区疼痛或压痛的原因。由于肝细胞坏死,释出细胞内的酶类入血,故血清谷丙转氨酶(SGPT)等升高,同时还可引起多种肝功能异常。肝细胞坏死较多时,胆红质的摄取、结合和分泌发生障碍,加之毛细胆管受压或有胆栓形成等则可引起黄疸。
结局:急性肝炎大多在半年内可逐渐恢复。点状坏死的肝细胞可完全再生修复。一部分病例(多为乙型、丙型肝炎)恢复较慢,需半年到一年,少数病例(约1%)可发展为慢性肝炎。极少数可恶化为重型肝炎。
2.慢性(普通型)肝炎 病毒性肝炎病程持续在一年(国外定为半年)以上者即为慢性肝炎。其中乙型肝炎占绝大多数(80%),也有近年明确的丙型肝炎。按病程、肝功能情况、免疫状态及病变等的不同将慢性肝炎分为持续性(迁延性)和活动性(进展性)二种。
(1)慢性持续性肝炎(chronic persistent hepatitis,CPH):临床症状常较轻或仅有肝功能异常。镜下,肝细胞变性、坏死较急性时减轻,Kupffer细胞增生活跃,汇管区或小叶内慢性炎性细胞浸润明显。有时汇管区可因有少量结缔组织增生而变宽。肝小叶轮子廓清楚,小叶界板无破坏。肉眼观,肝体积增大,但表面平滑。此型肝炎一般发展缓慢,经过较好,大多数可以恢复,少数可转变为慢性活动性肝炎。
(2)慢性活动性肝炎(chuonic active hepatitis,CAH):此型肝炎病变较重,肝功能持续异常。镜下,肝细胞变性坏死更为广泛而严重。肝细胞坏死呈灶状或条带状,并具有以下二种特征。①小叶周边的肝细胞界板受到破坏,界板肝细胞呈灶状坏死、崩解,伴有炎性细胞浸润,称为碎片状坏死(piecemeal necrosis)(图10-37);②小叶中央静脉与汇管区之间或两个中央静脉之间出现肝细胞坏死带,称桥接坏死(bridging necrosis)。坏死区可出现肝细胞不规则再生。小叶周边部坏死区纤维组织增生呈星芒状向小叶内伸展,并与小叶内肝细胞坏死处网状纤维支架塌陷而胶原化的纤维条索相连接,形成纤维间隔而分割小叶结构。肉眼观,在肿大的肝表面,上述纤维化明显区呈不平滑颗粒状,质地较硬。此型常见于乙、丙型肝炎,除肝外,患者还有脾肿大等全身改变,如不及时治愈大都转入肝硬变。

图10-37 慢性活动性肝炎
肝细胞明显气球样变和嗜酸性变,小叶界板破坏呈现碎片状坏死,门管区见炎性细胞浸润
毛玻璃样肝细胞 多见于HBsAg携带者及慢性肝炎患者的肝组织。光镜下,HE染色切片上,此等肝细胞浆内充满嗜酸性细颗粒状物质,不透明似毛玻璃样故称毛玻璃样肝细胞(图10-38)。这些细胞内含大量HBsAg,电镜下呈线状或小管状积存在内质网池内。用免疫酶标法或免疫荧光法可呈HBsAg阳性反应。(图10-39)。

图10-38 毛玻璃样肝细胞

图10-39 肝细胞内的乙型肝炎表面抗原
免疫酶标法(双PAP)染色显示肝细胞浆内的HBsAg
3.重量病毒性肝炎 本型病情严重。根据起病急缓及病变程度,可分为急性重型和亚急性重型二种。
(1)急性重型肝炎:少见。起病急,病变发展迅猛、剧烈,病死率高。临床上又称为暴发型、电击型或恶性型肝炎。
本型病变可见肝细胞坏死严重而广泛。肝索解离,肝细胞溶解,出现弥漫性的大片坏死。坏死多自小叶中央开始,向四周扩延,仅小叶周边部残留少数变性的肝细胞。肝窦明显扩张充血并出血,Kupffer细胞增生肥大,并吞噬细胞碎屑及色素。小叶内及汇管区有淋巴细胞和巨噬细胞为主的炎性细胞浸润(图10-40)。残留的肝细胞再生现象不明显。肉眼观,肝体积显著缩小,尤以左叶为甚,重量减至600~800g,质地柔软,表面被膜皱缩(图10-41)。切面呈黄色或红褐色,有的区域呈红黄相间的斑纹状,故又称急性黄色肝萎缩或急性红色肝萎缩。

图10-40 急性重型肝炎
肝细胞大片坏死消失,小叶中心部最重,周边部残存的肝细胞变性。坏死区有炎性细胞浸润

图10-41 急性重型肝炎
肝体积显著缩小,尤以左叶为著,表面被膜皱缩
临床病理联系及结局:由于大量肝细胞的迅速溶解坏死,可导致:①胆红质大量入血而引起黄疸(肝细胞性黄疸);②凝血因子合成障碍导致出血倾向;③肝功能衰竭,对各种代谢产物的解毒功能发生障碍。此外,由于胆红素代谢障碍及血循环障碍等,还可导致肾功能衰竭(肝肾综合征hepatorenal syndrome)。急性重型肝炎的死因主要为肝功能衰竭(肝昏迷),其次为消化道大出血或急性肾功能衰竭等。弥散性血管内凝血(DIC)也较常见,是引起严重出血、致死的另一个因素。
本型肝炎如能渡过急性期,部分病例可发展为亚急性型。
(2)亚急性重型肝炎:多数是由急性重型肝炎迁延而来或一开始病变就比较缓和呈亚急性经过。少数病例可能由普通型肝炎恶化而来。本型病程可达一至数月。
病变特点为,既有大片的肝细胞坏死,又有肝细胞结节状再生。由于坏死区网状纤维支架塌陷和胶原纤维化,致使再生的肝细胞失去原有的依托呈不规则的结节状,失去原有小叶的结构和功能。小叶内外有明显的炎性细胞浸润。小叶周边部小胆管增生并可有胆汁淤积形成胆栓。肉眼观,肝不同程度缩小,被膜皱隔,呈黄绿色(亚急性黄色肝萎缩)。病程长者可形成大小不等的结节,质地略硬。切面黄绿色(胆汁淤积),交错可见坏死区及小岛屿状再生结节。
此型肝炎如及时治疗有停止进展和治愈的可能。病程迁延较长(如1年)者,则逐渐过渡为坏死后性肝硬变。病情进展者可发生肝功能不全。
发病机制
病毒性肝炎的发病是病毒与机体之间相互作用的结果。病毒的量、毒力和侵入途径与发病有一定关系。小量病毒往往只引起隐性感染,而大量病毒则可导致严重的病变。肝炎的病变程度和类型与机体的免疫状态也有密切关系。现仅就研究较多的乙型肝炎的肝损伤及其不同类型的发病机制简述如下。
(1)肝细胞损伤的机制:乙型肝炎病毒侵入机体后进入肝细胞内复制繁殖,然后以发芽形式从肝细胞释出入血。在肝细胞表面则留下病毒抗原成分,此时并不引起明显的肝细胞损伤。病毒入血后,刺激机体免疫系统产生细胞免疫和体液免疫。前者通过杀伤性T细胞(K细胞)及NK细胞等的直接作用和抗体依赖性的细胞毒作用,后者通过产生各种特异性抗体,均能对血中病毒进行反应加以杀灭。但同时也能对受病毒感染的肝细胞(膜上含病毒抗原成分)进行攻击,使肝细胞受到破坏发生坏死。一般认为,T细胞介导的细胞免疫反应是病毒感染后引起肝细胞损伤的主要因素。近年研究表明,在细胞免疫反应中,靶细胞抗原不一定只是HBsAg,还可有肝细胞膜脂蛋白及HBsAg。
(2)乙型肝炎的发病机制:应用上述肝细胞免疫损伤机制可以解释乙型肝炎出现的不同类型:①T细胞功能正常,感染病毒量多,毒力强时受感染及免疫损伤的肝细胞多而重,表现为急性重型肝炎;②T细胞功能正常,病毒量较少,毒力较弱则发生急性普通型肝炎;③T细胞功能正常,病毒量甚少,毒力很弱则表现为轻型或亚临床型肝炎;④T细胞功能不足,免疫反应仅能清除部分病毒和损伤部分受感染的肝细胞,未清除的病毒可继续繁殖并感染,反复发生部分肝细胞损伤,结果表现为慢性肝炎;⑤机体免疫功能缺陷,T细胞呈免疫耐受状态,此时病毒与宿主共生。病毒在肝细胞内持续复制,感染的肝细胞也不受免疫损伤,此时则表现为无症状的病毒携带者。
四、酒精性肝病
酒精性肝病(alcoholic liver disease)为慢性酒精中毒的主要表现之一。长期大量酗酒者据统计有10%~20%发生此类损伤。严重时出现临床表现,如呕吐、呕血或黑便,其中部分可发生黄疸、肝功能衰竭。
病变
慢性酒精中毒主要引起肝的3种损伤,即脂肪肝、酒精性肝炎和酒精性肝硬变。三者可单独出现,也可同时并存或先后移行。一般认为脂肪肝在先,或经过酒精性肝炎再演变为肝硬变,或直接演变为肝硬变。
1.脂肪肝 酒精中毒最常见的肝病变是脂肪变性。肝细胞含有相当大的脂滴,可将胞核推挤到细胞一侧,肝细胞肿大变圆。小叶中央区受累明显,有时同时伴有各种程度的肝细胞水样变性。单纯的脂肪肝常无症状。
2.酒精性肝炎(alcoholic hepatitis) 在有临床肝症状表现的病例,常出现3种病变:肝细胞脂肪变性,酒精透明小体(alcoholic hyalin,AH 图10-42)形成和灶状肝细胞坏死伴中性粒细胞浸润。

图10-42 酒精性肝炎
图中央区肝细胞浆内见成群的酒精透明小体 ×400
3.酒精性肝硬变(alcoholic cirrhosis) 一般认为此种肝硬变是由脂肪肝和酒精性肝炎进展而来。一般的脂肪肝,如继续酗酒则多发展为酒精性炎,再演变为肝硬变。酒精性肝炎时肝细胞发生坏死,最终引起纤维化。相邻肝小叶的纤维化条索相互连接,导致肝小叶的正常结构被分割破坏,发展成假小叶及肝细胞结节状再生,形成酒精性肝硬变。
发病机制
肝是酒精代谢、降解的主要场所。酒精对肝有直接损伤作用。其机制如下:
1.NADH对NAD比值增高的效应 进入肝内的酒精,在乙醇脱氢酶和微粒体乙醇氧化酶系的作用下转变为乙醛,再转变为乙酸。后一反应使辅酶Ⅰ(NAD)转变为还原型辅酶Ⅰ(NADH),因而NADH/NAD值增高。①NADH增多有抑制线粒体三羧酸循环的作用,从而使肝细胞对脂肪酸的氧化能力降低,可引起脂肪在肝内堆积而发生脂肪肝。②NADH增多可使细胞代谢中的乳酸增多,后者对肝内脂肪变及胶原形成等有促进作用。③NADH增多,使线粒体增加以NADH的再氧化,致耗氧过多。肝细胞因缺氧而易于发生坏死和纤维化。
2.乙醛的毒性作用 在酒精代谢过程中产生的乙醛具有强烈的生化反应和毒性,可影响肝细胞膜的性状及抑制肝细胞合成的蛋白的分泌排出。
3.营养缺乏作用 嗜酒者常有的营养不足,尤其是蛋白质缺乏,使肝内氨基酸及酶类减少,可以促进酒精的毒性作用。
五、肝硬变
肝硬变(liver cirrhosis)是一种常见的慢性肝病,可由多种原因引起。肝细胞弥漫性变性坏死,继而出现纤维组织增生和肝细胞结节状再生,这三种改变反复交错进行,结果肝小叶结构和血液循环途径逐渐被改建,使肝变形、变硬而形成肝硬变。本病早期可无明显症状,后期则出现一系列不同程度的门静脉高压和肝功能障碍。
肝硬变按病因分类为:病毒肝炎性、酒精性、胆汁性、隐源性肝硬变。按形态分类为:小结节型、大结节型、大小结节混合型及不全分隔型肝硬变(为肝内小叶结构尚未完全改建的早期硬变)。我国常用的分类是结合病因及病变的综合分类,分为:门脉性、坏死后性、胆汁性、淤血性、寄生虫性和色素性肝硬变等。以上除坏死后性相当于大结节及大小结节混合型外,其余均相当于小结节型。其中门脉性肝硬变最常见,其次为坏死后性肝硬变。其它类型较少。
(一)门脉性肝硬变
门脉性肝硬变(portal cirrhosis),旧称雷奈克(Laennec)肝硬变,相当于小结节型肝硬变。为各型肝硬变中最常见者。本病在欧美因长期酗酒者引起多见(酒精性肝硬变),在我国及日本,病毒性肝炎则可能是其主要原因(肝炎后肝硬变)。
病因和发病机制
1.病毒性肝炎 慢性病毒性肝炎,尤以乙型慢性活动性肝炎是肝硬变的主要原因,其中大部分发展为门脉性肝硬变。在肝硬变患者肝内常显HBsAg阳性。其阳性率高达76.7%。另外,近年明确的丙型肝炎,大部可转为慢性,由慢性活动性肝炎转为肝硬变者达35%。
2.慢性酒精中毒 在欧美国家因长期酗酒引起的肝硬变可占总数的40%~50%。
3.营养缺乏 此项因素作为肝硬变的原因尚有争议。动物实验表明,饲喂缺乏胆碱或蛋氨酸食物的动物,可经过脂肪肝发展为肝硬变。
4.毒物中毒 某些化学毒物对肝有破坏作用,长期作用可引起肝硬变。临床上偶有因含砷的杀虫剂、辛可芬、四氯化碳、黄磷等慢性中毒引起肝硬变的报告。
上述各种因素首先引起肝细胞脂肪变、坏死及炎症等,以后在坏死区发生胶原纤维增生。后者主要来自增生的纤维母细胞、局部的贮脂细胞及因肝细胞坏死,局部的网状纤维支架塌陷,网状纤维融合形成胶原纤维(无细胞硬化)。初期增生的纤维组织虽形成小的条索但尚未互相连接形成间隔而改建肝小叶结构时,称为肝纤维化。为可复性病变,如果病因消除,纤维化尚可被逐渐吸收。如果继续进展,小叶中央区和汇管区等处的纤维间隔互相连接,终于使肝小叶结构和血液循环被改建而形成肝硬变(图10-43)。
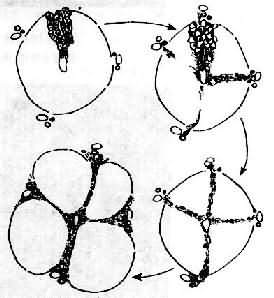
图10-43 门脉性肝硬变假小叶形成过程示意图
病变
肉眼观,早、中期肝体积正常或略增大,质地正常或稍硬。后期肝体积缩小 ,重量减轻,由正常的1500g减至1000g以下。肝硬度增加,表面呈颗粒状或小结节状,大小相仿,最大结节直径不超过1.0cm(图10-44)。切面见小结节周围为纤维组织条索包绕。结节呈黄褐色(脂肪变)或黄绿色(淤胆)弥漫分布于全肝。

图10-44 门脉性肝硬变
肝缩小变硬,表面呈弥漫的细颗粒状
镜下,正常肝小叶结构被破坏,由广泛增生的纤维组织将肝小叶分割包绕成大小不等、圆形或椭圆形肝细胞团,即假小叶(图10-45)。假小叶内肝细胞索排列紊乱,小叶中央静脉缺如、偏位或有两个以上,有时还可见被包绕进来的汇管区。再可见再生的肝细胞结节(也可形成假小叶),其特点是肝细胞排列紊乱,胞体较大,核大染色较深,常出现双核肝细胞。增生的纤维组织常压迫、破坏细小胆管,引起小胆管内淤胆。此外,还可见到新生的细小胆管和无管腔的假胆管(已用角蛋白单克隆抗体免疫组化技术证明,假胆管来源于胆管上皮)。
图10-45 门脉性肝硬变
图中央的肝细胞团由增生的结缔组织所包绕,为一个典型的假小叶
由于上述的肝细胞坏死、纤维组织增生和假小叶形成,相应地破坏并改建肝内肝血管系统,导致异常吻合枝的形成和血管网的减少。
临床病理联系
1.门脉高压症 是由于肝内血管系统在肝硬变时被破坏改建引起:①由于假小叶形成及肝实质纤维化的压迫使小叶下静脉(窦后)、小叶中央静脉及肝静脉窦受压,致门静脉的回流受阻。②肝动脉与门静脉间形成异常吻合支(图10-46),压力高的动脉血流入门静脉,使后者压力增高。
 
图10-46 肝硬变时肝内血液循环变化示意图
正常时肝内血液循环 肝硬变时肝内血管异常吻合
门脉压升高,胃、肠、脾等器官的静脉血回流受阻。晚期因代偿的调,临床出现:①脾肿大(splenomegaly),重量多在500g以下,少数可达800~1000g。镜下,红髓内有含铁血黄素沉着及纤维组织增生,形成黄褐色的含铁结节。②胃肠淤血,粘膜淤血、水肿,致患者食欲不振,消化不良。③腹水(ascites),在晚期出现,在腹腔内聚积大量淡黄色透明液体(漏出液)。腹水形成原因主要有:由于小叶中央静脉及小叶下静脉受压,肝窦内压上升,液体自窦壁漏出,部分经肝被膜漏入腹腔;肝细胞合成白蛋白功能降低,导致低蛋白血症,使血浆胶体渗透压降低;肝灭能作用降低,血中醛固酮、抗利尿素水平升高,引起水、钠潴留。④侧支循环形成,门静脉压升高使部分门静脉血经门体静脉吻合枝绕过肝直接回心(图10-47)。主要的侧支循环和合并症如:食管下段静脉丛曲张,如破裂可引起大呕血,是肝硬变患者常见的死因之一;直肠静脉丛曲张,破裂常发生便血,长期便血可引起贫血;脐周围静脉网曲张,临床上出现海蛇头(caput medusae)现象。

图10-47 肝硬变时侧支循环模式图
2.肝功能不全 由于肝实质长期反复受破坏,引起肝功能障碍。主要表现有:①睾丸萎缩,男子乳房发育症(可能因肝对雌激素灭能作用减弱);②蜘蛛状血管痣,为出现于体表的小动脉末梢扩张,也可能与体内雌激素过多有关;③出血倾向,患者有鼻衄、牙龈出血、粘膜、浆膜出血及皮下淤斑等。主要由于肝合成凝血因子及纤维蛋白原减少及脾肿大、功能亢进加强了对血小板的破坏;④肝细胞性黄疸,因肝细胞坏死,胆汁淤积而来,多见于肝硬变晚期;⑤肝性脑病(肝昏迷),是晚期肝功能衰竭引起的一种神经精神综合征,主要由于肠内含氮物质不能在肝内解毒而引起的氨中毒,为肝硬变患者常见的死因之一。
结局
肝硬变时肝组织已被增生的纤维组织改建,不易从形态结构上完全恢复正常,但是由于肝有强大代偿能力,只要及时治疗,常使疾病处于相对稳定状态,可维持相当长时期。此时肝细胞的变性、坏死基本消失,纤维母细胞的增生也可停止。但如病变持续进行,发展到晚期,肝功能衰竭,患者可因肝昏迷而死亡。此外,常见的死因还有食管下段静脉丛破裂引起的上消化道大出血,合并肝癌及感染等。
(二)坏死后性肝硬变
坏死后性肝硬变(postnecrotic cirrhosis),相当于大结节型肝硬变和大小结节混合型肝硬变,是在肝实质发生大片坏死的基础上形成的。
病因
1.肝炎病毒感染 现知大部为HBV感染,也有HBV与HDV复合感染。VEV的感染多在孕妇。患者多患亚急性重型肝炎,病程迁延数月至一年以上,则逐渐形成坏死后性肝硬变。另外,慢性活动性肝炎反复发作并且坏死严重时,也可引起。
2.药物及化学物质中毒。
病变
肉眼观,肝体积缩小,重量减轻,质地变硬。表面有较大且大小不等的结节,最大结节直径可达6cm。由于形成大小不等的结节常使肝变形(图10-48),肝左叶明显萎缩,右叶相对肥大隆起。

图10-48 坏死后性肝硬变
肝体积缩小,表面有大小不等的粗大结节
镜下,肝小叶呈灶状、带状甚至整个小叶坏死,代之以纤维组织增生,形成间隔,将原来的肝小叶分割为大小不等的假小叶。假小叶内肝细胞常有不同程度的变性和胆色素沉着。假小叶间的纤维间隔较宽阔且厚薄不均,其中炎性细胞浸润、小胆管增生均较显著,这些均与门脉型硬变不同。但本型病情进展较慢,病程较久者在病变上亦不易与门脉性硬变鉴别。
较高。
(三)胆汁性肝硬变
胆汁性肝硬变(billiary cirrhosis)是因胆道阻塞淤胆而引起的肝硬变,较少见,可分为继发性与原发性两类。原发性者更为少见。
1.继发性胆汁性肝硬变
病因:常见的原因为胆管系统的阻塞,如胆石、肿瘤(胰头癌、Vater壶腹癌)等对肝外胆道的压迫,引起狭窄及闭锁。在儿童患者多因肝外胆道先天闭锁,其次是总胆管的囊肿、囊性纤维化(cystic fibrosis)等。胆道系统完全闭塞6个月以上即可引起此型肝硬变。
病变:肝体积常增大,表面平滑或呈细颗粒状,硬度中等。肝外观常被胆汁染成深绿或绿褐色。
镜下,肝细胞胞浆内胆色素沉积,肝细胞因而变性坏死。坏死肝细胞肿大,胞浆疏松呈网状、核消失,称为网状或羽毛状坏死。毛细胆管淤胆、胆栓形成。胆汁外溢充满坏死区,形成胆汁湖。汇管区胆管扩张及小胆管增生。纤维组织增生使汇管区变宽、伸长,但在较长时期内并不侵入肝小叶内。故小叶的改建远较门脉性及坏死后性肝硬变为轻。伴有胆管感染时则见汇管区及增生的结缔组织内有多量中性粒细胞浸润甚至微脓肿形成。
2.原发性胆汁性肝硬变 本病又称慢性非化脓性破坏性胆管炎。很少见,多发生于中年以上妇女,男性患者不超过10%。临床表现为长期梗阻性黄疸、肝大和因胆汁刺激引起的皮肤瘙痒等。但肝内外的大胆管均无明显病变。本病还常伴有高脂血症和皮肤黄色瘤。
病因:不明,一般认为,可能与服用某些药物诱发肝胆管损伤及自向免疫反应有关。
病变:早期汇管区小叶间胆管上皮空泡变性及坏死并有淋巴细胞浸润,其后则有纤维组织的增生及胆小管的破坏、增生并出现淤胆现象。汇管区增生的纤维组织进而侵入肝小叶内,形成间隔,分割小叶最终发展为肝硬变。在汇管区有时可见铜的沉积,系因淤胆造成铜的肠肝循环障碍所致。
(四)其它类型肝硬变
淤血性肝硬变 本病见于慢性充血性心力衰竭。长期淤血缺氧,使肝小叶中央区肝细胞陷于萎缩、坏死,最后纤维化。如淤血持续存在,进而形成纤维条索分割肝小叶而形成肝硬变。
色素性肝硬变 多见于血色病(hemochromatosis)患者,由于肝内有过多的含铁血黄素沉着而形成。
寄生虫性肝硬变 主要见于慢性血吸虫病(详见寄生虫病)。
六、药物性肝损伤
进入体内的药物,无论是口服或注射均要经过肝代谢或解毒。某些药物可引起肝损伤。此种损伤系代谢过程中药物或其代谢产物直接对肝的毒性作用所致。损伤程度与药物毒性�
|