 |
 |
关键词:高铁血红蛋白血症;细胞色素还原酶;基因;点突变
摘要 目的 为了查明中国人遗传性高铁血红蛋白血症(RCM)患者的NADH-细胞色素b5还原酶(b5R)基因突变类型,探讨RCM发生的分子机制。方法 从已报道的1例RCMⅠ型患者的外周血白细胞中提取RNA,应用逆转录-聚合酶链反应法(RT-PCR)扩增了b5RcDNA(921bp),测定其b5R cDNA的全部编码序列。结果 b5R cDNA基因的第203位密码子存在(TGC→TAC)Cys→Tyr的错义突变。经基因组PCR-限制性片段长度多态性(RFLP)分析证实该突变并非PCR过程中的错配。结论 Cys203→Tyr是RCM的一种新的基因突变类型。
A novel point mutation inNADH-cytochrome b5 reductase gene
WANG Yao* ,WU Yushui,YANG Wenxi,et al.*
Center for Medical Laboratory,Fuzhou General Hospital,Fuzhou 350025
Abstract Objective To characterize theb5R gene mutation in a Chinese patient with recessive congenital methemoglobinemia type Ⅰ(RCM Ⅰ).Methods Total RNA was extracted from the peripheralleukocytes of the patient and cDNA was synthesized by RT-PCR. The coding region of b5RcDNA (921bp) was analysed by sequencing of the RT-PCR products.Results andConclusion A novel mutation of Cys203(TGC)→Try(TAC) in exon 7 wasidentified,which was further confirmed by restriction enzyme analysis of the genomic DNAfragment.
Key words Methemoglobinemia Cytochrome reductase Gene Pointmutation
遗传性高铁血红蛋白血症(RCM),是一种以发绀为主要临床表现的常染色体隐性遗传病,其病因是NADH-细胞色素b5还原酶(b5R)先天缺陷。近年来对该病的分子生物学研究表明,其发病的分子基础主要是由于点突变引起的酶蛋白结构改变,导致稳定性下降以及酶活力降低。该病在我国并不罕见。近几年来,我们已在福州周围地区发现5个家系9例患者,并对这些患者进行了生物化学、免疫学、分子生物学的研究,发现了两种突变类型(Arg57→Glu[1] 和Leu72→Pro[2] ),其中Leu72→Pro为世界首报。最近我们又对浙江舟山1个家系的患者进行了基因结构分析,发现为一种新的突变类型(Cys203→Tyr),现报道其结构与结果。
病例和方法
1 先证者,女,74岁,浙江省舟山市人。临床表现为明显发绀,无呼吸困难和神经系统症状,经血液生化检测,高铁血红蛋白含量为19%,b5R活性显著降低。患者之父及一兄一弟也自幼发绀,先证者的一个女儿为杂合子。该家系曾于1984年由杨文西等[3] 作过报道,诊断为RCMⅠ型,并作过家系调查。
2 主要试剂与仪器
限制性内切酶EcoRⅠ、BamHⅠ、RsaⅠ以及T4DNA连接酶、逆转录试剂盒、Φ×174DNA/HaeⅢmarker均为Promega公司产品。RNA和DNA提取试剂盒购自Qiagen公司;测序试剂盒及373A荧光自动DNA序列分析仪为ABI公司产品;Taq酶及聚合酶链反应(PCR)扩增试剂盒为上海Sangon公司产品。PE2400扩增仪购自PE公司。
3 引物设计与合成
依据b5R cDNA的编码序列 ,其引物设计为:
上游P1 为5′-GGGAATTCATGGGGGCCCAGCTC-AGCACG-3′,5′端含EcoRⅠ酶切位点接头;
下游P2 为5′-GGGGATCCCCTCAGAAGACGA-AGCAGCGC-3′,5′端含BamHⅠ酶切位点接头。
扩增片段长度为921bp。以上引物均由中国科学院上海植物生理研究所合成。
4 方法
4.1 b5R cDNA的克隆及序列分析:先证者和正常人淋巴细胞总RNA提取按Qiagen试剂盒说明书的方法进行。取RNA1~2μg以oligod T18为引物,按Promega公司逆转录试剂盒说明书的方法合成cDNA第1条链。取4μlcDNA产物作为模板,以引物P1 和P2 按常规PCR方法进行。
4.2 PCR产物克隆测序:回收并纯化PCR产物,用EcoRⅠ和BamHⅠ酶切后克隆到pWR450-1载体上(复旦大学遗传学研究所惠赠),酶切鉴定重组子。荧光标记P1 和P2 引物,测定阳性克隆DNA序列。
4.3 基因组DNA片段的扩增和限制性酶切图谱分析:先证者及其女儿的基因组DNA提取按Qiagen公司DNA提取试剂盒的说明书进行。分别以患者和正常人DNA为模板,用下列引物进行PCR扩增:
T1 :5′-AGTACACCTGGGCGGGGCGG-3′,
T2 : 5′-AGAATCTAGCAGAGCTGTGCA-3′。
扩增片段长度为262bp。PCR产物用RsaⅠ 10U酶切,120g/L聚丙烯酰胺凝胶电泳(PAGE)分离,紫外灯下观察结果。
结 果
1 b5R cDNA的克隆和序列分析
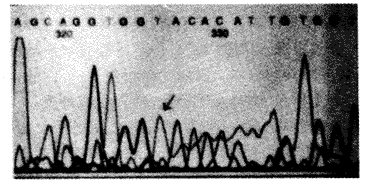
与正常人比较,先证者在第7外显子的第203号密码子上存在TGC→TAC点突变(见图1),即造成氨基酸残基Cys→Tyr的替换。
箭头所指为第7外显子的第203位密码子碱基G→A
(Cys203→Tyr)
图1 先证者b5R cDNA基因序列分析
2 点突变(CD203TGC→TAC)基因诊断结果
由于第203密码子突变后导致该位点产生一个RsaⅠ酶切位点,将患者和正常人第7外显子PCR扩增产物用RsaⅠ消化后,PAGE结果显示正常人产生259bp一个条带,RCM患者纯合子则产生134bp、125bp二个条带,杂合子可见259bp、134bp和125bp三个条带(见图2),两种基因型得以准确区分。经PCR-限制性片段长度多态性(PCR-RFLP)分析,先证者为此突变的纯合子,其女儿为杂合子。
M:Φ×174DNA/Hae Ⅲ Marker;1:先证者的女儿,为杂合子,出现259bp、134bp和125bp三条带;2:先证者为纯合子,出现134bp和125bp二条带;3:正常对照,仅有259bp条带
图2 PCR-RFLP分析图谱
讨 论
广义的RCM是指所有由于遗传性缺陷造成的血中高铁血红蛋白含量增高的疾患,包括b5R缺陷,细胞色素b5缺陷,血红蛋白M(HbM)血症和部分不稳定血红蛋白病。其中以b5R缺陷较为多见,故当前在国外文献中,如不作特别说明(例如指明为HbM所致),一般RCM即专指遗传性b5R缺陷症。
自1845年Francois报道了第1例RCM以来,文献已报道了500余例。此病的临床表现主要为发绀,严重者可有头痛、易疲乏、劳累性呼吸困难以及神经、精神症状。根据临床表现和组织中酶活性分布不同,通常将本病分为三型:Ⅰ型(红细胞型)最为多见,酶活性降低仅限于红细胞。Ⅱ型(全身细胞型)较少见,全身细胞酶活力均下降,临床上除有发绀外,尚有精神发育迟缓和一系列神经、精神症状,患者常于幼年期夭折。Ⅲ型(血细胞型)最为少见,酶活力下降局限于白细胞、红细胞和血小板等血细胞。但根据近来的文献报道,以往报道的所谓Ⅲ型实际上与Ⅰ型是同一类型[4] 。
人b5R cDNA全长1974bp,其中903bp编码301个氨基酸残基[5] 。迄今所发现的该病的分子异常主要是由于基因编码序列点突变及缺失导致酶活性改变所致,而基因的调控序列尚未发现异常。不同表型的RCM其b5R基因的突变位点不同。迄今为止已发现了15种突变表型:在Ⅰ型中有5种点突变(Arg57→Gln[1] , Val 105→Met, Leu 148→Pro[6] , Glu 212→Lys[7] ,Leu 72→Pro[2] );在Ⅱ型中有5种点突变(Ser 127→Pro, Arg 218Stop,Cys 203→Arg[8] , Pro 95→His和Thr42Stop)和两种3bp缺失及第6外显子的缺失(delPhe-298、del Met-272、del exon 6)以及两种剪切位点突变(5′splice site of exon5和3′splice site of exon 7)。在此15种突变类型中有5种属于Ⅰ型患者,突变影响的基因近5′端;在Ⅱ型中的大部分突变基因位于近3′端。这两个区域大致分别同酶蛋白与FAD及NAD的结合区域相一致。对Ⅰ型患者突变酶的功能研究表明,酶蛋白分子不稳定(易降解)是主要原因,而酶活性相对正常。由于红细胞已失去继续合成b5R的能力,且寿命长达120天,所以主要表现为红细胞中b5R显著缺乏,而其他组织细胞因能继续不断合成新的b5R,故虽b5R降低较快,但能不断补充,对其他组织细胞影响较少,临床主要表现为血中高铁血红蛋白升高,出现发绀。而Ⅱ型突变酶似乎主要影响酶的催化活性,对全身细胞均产生严重影响。除了高铁血红蛋白升高所致发绀外,还有脑神经脂类合成障碍,造成严重的神经、精神症状。
在本研究中先证者的b5R基因中存在Cys203→Tyr(TGC→TAC)的突变,为世界上首次报道。1995年Vieira等[8] 曾报道Ⅱ型患者有Cys203→Arg(TGC→GGC)突变,其突变位点与本文中的患者相同,但替换的氨基酸却不同,带负电荷的半胱氨酸被带正电荷的精氨酸所置换,由此造成的临床表型也不相同。在同一位点发生不同的氨基酸突变而造成不同的临床表型这一现象,在RCM中是首次报道。从这个现象我们可推测:①Ⅰ型患者的突变位点并不总发生在近基因的5′端,也可位于近基因的3′端。Mary等[7] 也曾报道在近3′端点突变Glu212→Lys,临床表现为Ⅰ型的报道;②Ⅰ和Ⅱ型患者均可在第203位密码子发生突变,此位点可能是一突变热点;③按Chou等的方法[9] 由氨基酸序列推导的肽链二级结构,本例突变位点在b5R二级结构中位于β2 -片层与NAD结合区,是b5R蛋白羧基端四个半胱氨酸中的一个。Cys203→Tyr突变引入了一个带苯酚侧链的氨基酸而取代了带巯基的半胱氨酸,虽然巯基侧链与苯酚侧链同为带负电荷的侧链,其pK3值相近(前者pK3为10.28,后者pK3为10.07),但因为带巯基的Cys对维持b5R的活性十分重要[10] ,所以引起b5R活性显著下降。
基金项目: 本课题受全军“八五”重点攻关课题[(159)-C10]资助
参考文献
1 黄长晖,谢毅,王瑶,等.中国人遗传性高铁血红蛋白血症二个家系所见Cytb5R基因Arg 57→Gln突变.中华血液学杂志,1997,18:200-203.
2 吴玉水,黄长晖,朱忠勇,等.中国人遗传性高铁血红蛋白血症NADH-细胞色素b5还原酶基因L72P突变.中华血液学杂志,1998,19:195-197.
3 杨文西,郑祖纽,方国安.Ⅰ型高铁血红蛋白还原酶缺乏症:附一个家系调查.舟山医学,1984,18:18-23.
4 Nagai T,Shirabe K,Yubisui T,et al.Analysis of mutant NADH-Cytochrome b5 reductase ″typeⅢ″methemoglobinemia can be explained as typeⅠwith an unstable reductase. Blood,1993,81:808-814.
5 Yubisui T,Naitoh Y,Zenno S,et al.Molecular cloning of cDNAs of human liver andplacenta NADH-cytochrome b5 reductase.Proc Natl Acad Sci U S A,1987,84:3609-3613.
6 Katsube T,Sakamoto N,Kobayashi Y,et al.Exonic point mutations in NADH-cytochrome b5reductase genes of homozygotes for hereditary methemoglobinemia.TypeⅠand Ⅲ:putativemechanisms of tissue-dependent enzyme deficiency.Am J Hum Genet,1991,48:799-807.
7 Mary MJ,Josef TP. A novel mutation found in the 3′domain of NADH-cytochrome b5reductase in an African-American family with typeⅠ congenitalmethemoglobinemia.Blood,1996,187:2993-2999.
8 Vieira LM,Kaplan JC,Kahn A,et al.Four new mutations in the NADH-cytochrome b5reductasegene from patients with reccessive congenital methemoglobinemia typeⅡ.Blood,1995,85:2254-2262.
9 Chou PY,Fasman GD.Empirical predictions of protein conformation.Ann RevBiochem,1978,47:251-278.
10 Shirabe K, Yubisui T,Borgese N,et al.Enzymatic instability of NADH-cytochrome b5reductase as a cause of hereditary methemoglobinemia type Ⅰ (red cell type).J BiolChem,1993,267:20414-20418.
收稿:1998-12-07
修回:1999-02-25, http://www.100md.com