紫杉醇的半合成
作者:王九一1 阎家麒
单位:王九一1 河南省分析测试中心,郑州 450002;阎家麒 河南绿十字生物工程研究所,郑州 450008
关键词:紫杉醇;10-脱乙酰巴卡亭Ⅲ;N-苯甲酰-(2R,3S)-3-苯基异丝氨酸;半合成
中国药物化学杂志990114 摘 要 以10-脱乙酰巴卡亭Ⅲ为起始原料,将其转化成7-三乙基硅烷10-脱乙酰巴卡亭Ⅲ,然后乙酰化成7-三乙基硅烷巴卡亭Ⅲ,用市售的紫杉醇侧链N-苯甲酰-(2R,3S)-3-苯基异丝氨酸在DCC和DAMP作用下与其偶联,用0.5%盐酸除去保护基团得紫杉醇.
Semisynthesis of Taxol
Wang Jiuyi
, http://www.100md.com
(He′nan Analysis and Testing Center,Zhengzhou 450002)
Abstract The starting material was 10-deacetylbaccatin Ⅲ,and this was converted to 7-triethylsilyl 10-deacetylbaccatin Ⅲ and then acetylated to give 7-triethylsilyl baccatin Ⅲ,which was then coupled with the commercially available side chain N-benzoyl-(2R,3S)-phenylisoserine in the presence of DCC and DMAP.Removal of the protecting group with 0.5% HCl gave Taxol in high yield.
, http://www.100md.com Key words Taxol;10-deacetylbaccatin Ⅲ;N-benzoyl-(2R,3S)-phenylisoserine;semisynthesis
紫杉醇(paclitaxel,商品名Taxol,1)是由红豆杉属植物分离提取的一种具有独特抗癌活性的紫杉烷二萜类化合物.它已在40多个国家获准上市,用于治疗晚期卵巢癌和乳腺癌等.目前其临床适应症扩大到肺癌、头颈部癌、软组织癌、白血病和胃肠道癌,被认为是迄今人类发现的最有效的抗癌药物〔1〕.目前,紫杉醇主要来源于几种红豆杉属植物的树皮,其含量约0.01%.近年来,化学家们对紫杉醇的化学全合成研究取得了可喜的进展〔2,3〕,但是,全合成路线复杂,成本过高,尚无实用价值.作者在紫杉醇的分离纯化研究中〔4,5〕,得到一种副产物:10-脱乙酰巴卡亭Ⅲ(10-deacetylbaccatin Ⅲ,2),它是半合成紫杉醇的主要原料,其得率高于目的物紫杉醇.最近有报道,从中国松科马尾松(Pinus massoniana Lamb.)和粗榧科中国粗榧(Cephalotaxus sinensis Rehd. et Wils.)的树叶中分离提取到化合物(2)〔6〕.这两种植物在我国资源丰富,蕴藏量数倍于红豆杉,从而为紫杉醇的半合成提供了较充足的原料.
, http://www.100md.com
1 合成路线设计
参照Denis合成路线〔7〕,使用N-苯甲酰-(2R,3S)-3-苯基异丝氨酸[N-benzoyl-(2R,3S)-3-phenylisoserine,3]这种市售的紫杉醇侧链对化合物(2)的13位羟基进行酯化.由于化合物(2)中的7,10,13位羟基活性不同,先用氯化三乙基硅烷保护7位羟基,得到7-三乙基硅烷-10-脱乙酰巴卡亭Ⅲ(4),再乙酰化10位羟基,得到7-三乙基硅烷巴卡亭Ⅲ(5).然后用侧链化合物(3)使13位羟基酯化,再在含水盐酸存在下,水解去除7位上的硅烷保护基团,得到紫杉醇.这样,只需3步反应即得到目标物.简化了合成步骤,提高了收率.合成路线如图1.
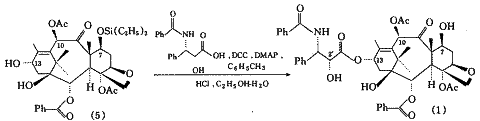
Fig.1 Route of synthesis
, 百拇医药
2 实验部分
2.1 7-三乙基硅烷10-脱乙酰巴卡亭Ⅲ(4)的制备
取化合物(2)1.0 g(1.8 mmol)溶于无水吡啶100 mL,通氮气,搅拌,缓慢滴加三乙基氯硅烷6.7 mL(6.0 g,40 mmol),将黄色反应液在0℃下搅拌24 h,加乙酸乙酯和水各200 mL,搅拌后静置分层,有机层经饱和硫酸铜溶液洗涤(300 mL×3),直至将过量的吡啶全部除去,再用水及饱和氯化钠100 mL洗涤.然后通过无水硫酸钠干燥,过滤后减压除去溶剂,浓缩物经230~400目硅胶-60(Merck)色谱柱纯化,用二氯甲烷-甲醇(v∶v=99∶1)洗脱,得化合物(4)1.12 g,收率:79%.mp 256~257℃(二氯甲烷-戊烷),[α]23D-23.6°(c=0.41,甲醇).1H-NMR δ:0.48~0.70(m,6H),0.94(t,J=8 Hz,9H),1.08(s,6H),1.57(s,1H),1.74(s,3H),1.86~1.95(m,1H),2.02(d,J=5 Hz,1H),2.09(d,J=1.1 Hz,3H),2.25~2.35(m,2H),2.28(s,3H),2.42~2.52(m,1H),3.95(d,J=7 Hz,1H),4.24(ABq,JAB=8.4 Hz,δA-δB=42,2H),4.31(d,J=2 Hz,1H),4.41(dd,J=6.6和10.6 Hz,1H),4.85~4.90(m,1H),4.95(dd,J=1.9和9.6 Hz,1H),5.17,(d,J=2 Hz,1H),5.60(d,J=7 Hz,1H),7.44~7.50(m,2H),7.57~7.62(m,1H),8.08~8.11(m,2H),与文献〔8〕值相符.
, 百拇医药
2.2 7-三乙基硅烷巴卡亭Ⅲ(5)的制备
将化合物(4)1.0 g(1.5 mmol)溶于无水吡啶43 mL,再滴加乙酰氯0.6 mL,在0℃下继续搅拌20 h.加乙酸乙酯和水各50 mL,搅拌,静置分层后,用乙酸乙酯(30 mL×2)提取水相.合并有机相,用饱和硫酸铜溶液洗涤至吡啶完全除去.然后依次用水及饱和氯化钠溶液洗涤,通过无水硫酸钠干燥.过滤后减压浓缩,浓缩物上230~400目硅胶-60(Merck)色谱柱,用二氯甲烷-甲醇(v∶v=99∶1)洗脱,得化合物(5)1.03 g,收率:86%.mp 253~254℃(二氯甲烷-戊烷),[α]23D-49°(c=0.4,甲醇).1H-NMR δ:0.52~0.65(m,6H),0.93(t,J=8 Hz,9H),1.04(s,3H),1.20(s,3H),1.61(s,1H),1.68(s,3H),1.83~1.92(m,1H),2.01(d,J=5 Hz,1H),2.17(s,3H),2.19(d,J=1.1 Hz,3H),2.24~2.28(m,2H),2.29(s,3H),2.41~2.58(m,1H),3.88(d,J=7 Hz,1H),4.23(AB q,JAB=8.1 Hz,δA-δB=45,2H),4.49(dd,J=6.6和10.5 Hz,1H),4.84(m,1H),4.96(d,J=9.6 Hz,1H),5.63(d,J=7 Hz,1H),6.46(s,1H),7.45~7.50(m,2H),7.57~7.63(m,1H),8.09~8.12(m,2H),与文献〔8〕值相符.
, 百拇医药
2.3 紫杉醇(1)的合成
将化合物(5)1.0 g(1.4 mmol)溶于无水甲苯70 mL,搅拌下加入化合物(3)1.3 g(4.6 mmol),再加入DCC 470 mg(4.5 mmol),反应5 min后加入DMAP 350 mg(2.8 mmol),搅拌4 min,加热至75℃,反应10 h.冷却后用适量乙酸乙酯稀释,静置分出有机层,依次用饱和碳酸氢钠溶液洗涤3次,用水洗涤2次,用饱和氯化钠溶液洗涤2次,然后用无水硫酸钠干燥.过滤后减压浓缩,硅胶色谱柱纯化,得C-7保护的紫杉醇衍生物.将其溶于预冷0℃的含0.5%盐酸的乙醇62 mL,0℃下通氮气搅拌30 h(用TLC检识至衍生物完全消失,反应即终止).反应液用乙酸乙酯450 mL稀释,用水洗涤,静置,分出有机层,再用水洗涤5次,用饱和氯化钠溶液洗涤2次,然后通过无水硫酸钠干燥.过滤后,滤液减压浓缩,上230~400目硅胶-60(Merck)色谱柱,用二氯甲烷-甲醇(v∶v=90∶10)洗脱,含水甲醇中结晶,得紫杉醇922 mg,收率:89%.mp 194~195℃,[α]24D-49.7°(c=0.36,甲醇).1H-NMR δ:1.14(s,3H),1.24(s,3H),1.69(s,3H),1.79(s,3H),1.83(s,1H),1.83~1.93(m,1H),2.23(s,3H),2.28~2.39(m,2H),2.38(s,3H),2.48(d,J=4 Hz,1H),2.48~2.57(m,1H),3.55(d,J=5 Hz,1H),3.79(d,J=7 Hz,1H),4.25(AB q,JAB=8.4 Hz,δA-δB=32,2H),4.37~4.44(m,1H),4.80(dd,J=2.5和5 Hz,1H),4.94(bd,J=8.0 Hz,1H),5.67(d,J=7 Hz,1H),5.78(dd,J=2.5和8.8 Hz,1H),6.23(t,J=9 Hz,1H),6.27(s,1H),7.01(d,J=8.9 Hz,1H),7.33~7.54(m,10H),7.59~7.64(m,1H),7.22~7.75(m,2H),8.12~8.15(m,2H).13C-NMR δ:9.57(CH3),14.83(CH3),20.83(CH3),21.62(CH3),22.85(CH3),26.69(CH3),35.65(CH3),35.73(CH3),43.20(C),45.86(CH),55.05(CH),58.67(C),72.20(CH),72.41(CH),73.23(CH),75.00(CH),75.58(CH),76.58(CH2),79.10(C),81.21(C),84.42(CH),127.06(CH),128.38(CH),128.72(CH),129.04(CH),129.21(C),130.22(CH),131.37(CH),133.26(C),133.71(CH),138.03(C),141.98(C),167.04(C),170.17(C),171.22(C),172.73(C),203.62(C).与文献〔9〕值相符,即与红豆杉提取纯化的紫杉醇结构一致.半合成紫杉醇纯度测定依据HPLC法〔10〕,其纯度为99.6%.
, http://www.100md.com
1通讯联系人
参考文献
[1] Kingston DGI,Molinero AA,Rimoldi JM.The taxane diterpenoids.In:Herz W,Kirby GW,Moore RE,et al. eds,Progress in the Chemistry of Organic Natural Products,Vol.61,New York:Spiringer-Verlag,1993.31~60
[2] Holton RA,Somoza C,Kim HB, et al.First total synthesis of taxol.J Am Chem Soc,1994,116(14):1579~1602
[3] Nicolaou KC,Yang Z,Liu JJ,et al.Total synthesis of taxol.Nature,1994,367(17):630~632
, 百拇医药
[4] 阎家麒,刘虹,王九一,等.东北红豆杉原料紫杉醇的提取及测定.中国医药工业杂志,1994,25(10):433~436
[5] 阎家麒,范金城,王九一.紫杉醇提取纯化工艺.中国医药工业杂志,1996,27(12):531~534
[6] 陈冲,罗思齐.10-脱乙酰浆果杉亭-Ⅲ的新药源研究.中国医药工业杂志,1997,28(8):375~376
[7] Denis JN,Greene AE.A highly efficient practical approach to natural taxol. Am Chem Soc,1988,110(17):5917~5919
[8] 岳琴,方起程,梁晓天.紫杉醇的半合成.药学学报,1996,31(12):911~917
[9] Chmurny GN,Hilton BD,Brobst S,et al.1H-and 13C-NMR assignments for taxol,7-epitaxol and cephalomannine.J Nat Prod,1992,55(4):414~423
[10] 阎家麒,周海龙,周龙云,等.紫杉醇原料药纯度的RP-HPLC测定.中国医药工业杂志,1996,27(10):455~457
收稿日期:1998-10-24, http://www.100md.com(王九一1 阎家麒)
单位:王九一1 河南省分析测试中心,郑州 450002;阎家麒 河南绿十字生物工程研究所,郑州 450008
关键词:紫杉醇;10-脱乙酰巴卡亭Ⅲ;N-苯甲酰-(2R,3S)-3-苯基异丝氨酸;半合成
中国药物化学杂志990114 摘 要 以10-脱乙酰巴卡亭Ⅲ为起始原料,将其转化成7-三乙基硅烷10-脱乙酰巴卡亭Ⅲ,然后乙酰化成7-三乙基硅烷巴卡亭Ⅲ,用市售的紫杉醇侧链N-苯甲酰-(2R,3S)-3-苯基异丝氨酸在DCC和DAMP作用下与其偶联,用0.5%盐酸除去保护基团得紫杉醇.
Semisynthesis of Taxol
Wang Jiuyi
, http://www.100md.com
(He′nan Analysis and Testing Center,Zhengzhou 450002)
Abstract The starting material was 10-deacetylbaccatin Ⅲ,and this was converted to 7-triethylsilyl 10-deacetylbaccatin Ⅲ and then acetylated to give 7-triethylsilyl baccatin Ⅲ,which was then coupled with the commercially available side chain N-benzoyl-(2R,3S)-phenylisoserine in the presence of DCC and DMAP.Removal of the protecting group with 0.5% HCl gave Taxol in high yield.
, http://www.100md.com Key words Taxol;10-deacetylbaccatin Ⅲ;N-benzoyl-(2R,3S)-phenylisoserine;semisynthesis
紫杉醇(paclitaxel,商品名Taxol,1)是由红豆杉属植物分离提取的一种具有独特抗癌活性的紫杉烷二萜类化合物.它已在40多个国家获准上市,用于治疗晚期卵巢癌和乳腺癌等.目前其临床适应症扩大到肺癌、头颈部癌、软组织癌、白血病和胃肠道癌,被认为是迄今人类发现的最有效的抗癌药物〔1〕.目前,紫杉醇主要来源于几种红豆杉属植物的树皮,其含量约0.01%.近年来,化学家们对紫杉醇的化学全合成研究取得了可喜的进展〔2,3〕,但是,全合成路线复杂,成本过高,尚无实用价值.作者在紫杉醇的分离纯化研究中〔4,5〕,得到一种副产物:10-脱乙酰巴卡亭Ⅲ(10-deacetylbaccatin Ⅲ,2),它是半合成紫杉醇的主要原料,其得率高于目的物紫杉醇.最近有报道,从中国松科马尾松(Pinus massoniana Lamb.)和粗榧科中国粗榧(Cephalotaxus sinensis Rehd. et Wils.)的树叶中分离提取到化合物(2)〔6〕.这两种植物在我国资源丰富,蕴藏量数倍于红豆杉,从而为紫杉醇的半合成提供了较充足的原料.
, http://www.100md.com
1 合成路线设计
参照Denis合成路线〔7〕,使用N-苯甲酰-(2R,3S)-3-苯基异丝氨酸[N-benzoyl-(2R,3S)-3-phenylisoserine,3]这种市售的紫杉醇侧链对化合物(2)的13位羟基进行酯化.由于化合物(2)中的7,10,13位羟基活性不同,先用氯化三乙基硅烷保护7位羟基,得到7-三乙基硅烷-10-脱乙酰巴卡亭Ⅲ(4),再乙酰化10位羟基,得到7-三乙基硅烷巴卡亭Ⅲ(5).然后用侧链化合物(3)使13位羟基酯化,再在含水盐酸存在下,水解去除7位上的硅烷保护基团,得到紫杉醇.这样,只需3步反应即得到目标物.简化了合成步骤,提高了收率.合成路线如图1.
Fig.1 Route of synthesis
, 百拇医药
2 实验部分
2.1 7-三乙基硅烷10-脱乙酰巴卡亭Ⅲ(4)的制备
取化合物(2)1.0 g(1.8 mmol)溶于无水吡啶100 mL,通氮气,搅拌,缓慢滴加三乙基氯硅烷6.7 mL(6.0 g,40 mmol),将黄色反应液在0℃下搅拌24 h,加乙酸乙酯和水各200 mL,搅拌后静置分层,有机层经饱和硫酸铜溶液洗涤(300 mL×3),直至将过量的吡啶全部除去,再用水及饱和氯化钠100 mL洗涤.然后通过无水硫酸钠干燥,过滤后减压除去溶剂,浓缩物经230~400目硅胶-60(Merck)色谱柱纯化,用二氯甲烷-甲醇(v∶v=99∶1)洗脱,得化合物(4)1.12 g,收率:79%.mp 256~257℃(二氯甲烷-戊烷),[α]23D-23.6°(c=0.41,甲醇).1H-NMR δ:0.48~0.70(m,6H),0.94(t,J=8 Hz,9H),1.08(s,6H),1.57(s,1H),1.74(s,3H),1.86~1.95(m,1H),2.02(d,J=5 Hz,1H),2.09(d,J=1.1 Hz,3H),2.25~2.35(m,2H),2.28(s,3H),2.42~2.52(m,1H),3.95(d,J=7 Hz,1H),4.24(ABq,JAB=8.4 Hz,δA-δB=42,2H),4.31(d,J=2 Hz,1H),4.41(dd,J=6.6和10.6 Hz,1H),4.85~4.90(m,1H),4.95(dd,J=1.9和9.6 Hz,1H),5.17,(d,J=2 Hz,1H),5.60(d,J=7 Hz,1H),7.44~7.50(m,2H),7.57~7.62(m,1H),8.08~8.11(m,2H),与文献〔8〕值相符.
, 百拇医药
2.2 7-三乙基硅烷巴卡亭Ⅲ(5)的制备
将化合物(4)1.0 g(1.5 mmol)溶于无水吡啶43 mL,再滴加乙酰氯0.6 mL,在0℃下继续搅拌20 h.加乙酸乙酯和水各50 mL,搅拌,静置分层后,用乙酸乙酯(30 mL×2)提取水相.合并有机相,用饱和硫酸铜溶液洗涤至吡啶完全除去.然后依次用水及饱和氯化钠溶液洗涤,通过无水硫酸钠干燥.过滤后减压浓缩,浓缩物上230~400目硅胶-60(Merck)色谱柱,用二氯甲烷-甲醇(v∶v=99∶1)洗脱,得化合物(5)1.03 g,收率:86%.mp 253~254℃(二氯甲烷-戊烷),[α]23D-49°(c=0.4,甲醇).1H-NMR δ:0.52~0.65(m,6H),0.93(t,J=8 Hz,9H),1.04(s,3H),1.20(s,3H),1.61(s,1H),1.68(s,3H),1.83~1.92(m,1H),2.01(d,J=5 Hz,1H),2.17(s,3H),2.19(d,J=1.1 Hz,3H),2.24~2.28(m,2H),2.29(s,3H),2.41~2.58(m,1H),3.88(d,J=7 Hz,1H),4.23(AB q,JAB=8.1 Hz,δA-δB=45,2H),4.49(dd,J=6.6和10.5 Hz,1H),4.84(m,1H),4.96(d,J=9.6 Hz,1H),5.63(d,J=7 Hz,1H),6.46(s,1H),7.45~7.50(m,2H),7.57~7.63(m,1H),8.09~8.12(m,2H),与文献〔8〕值相符.
, 百拇医药
2.3 紫杉醇(1)的合成
将化合物(5)1.0 g(1.4 mmol)溶于无水甲苯70 mL,搅拌下加入化合物(3)1.3 g(4.6 mmol),再加入DCC 470 mg(4.5 mmol),反应5 min后加入DMAP 350 mg(2.8 mmol),搅拌4 min,加热至75℃,反应10 h.冷却后用适量乙酸乙酯稀释,静置分出有机层,依次用饱和碳酸氢钠溶液洗涤3次,用水洗涤2次,用饱和氯化钠溶液洗涤2次,然后用无水硫酸钠干燥.过滤后减压浓缩,硅胶色谱柱纯化,得C-7保护的紫杉醇衍生物.将其溶于预冷0℃的含0.5%盐酸的乙醇62 mL,0℃下通氮气搅拌30 h(用TLC检识至衍生物完全消失,反应即终止).反应液用乙酸乙酯450 mL稀释,用水洗涤,静置,分出有机层,再用水洗涤5次,用饱和氯化钠溶液洗涤2次,然后通过无水硫酸钠干燥.过滤后,滤液减压浓缩,上230~400目硅胶-60(Merck)色谱柱,用二氯甲烷-甲醇(v∶v=90∶10)洗脱,含水甲醇中结晶,得紫杉醇922 mg,收率:89%.mp 194~195℃,[α]24D-49.7°(c=0.36,甲醇).1H-NMR δ:1.14(s,3H),1.24(s,3H),1.69(s,3H),1.79(s,3H),1.83(s,1H),1.83~1.93(m,1H),2.23(s,3H),2.28~2.39(m,2H),2.38(s,3H),2.48(d,J=4 Hz,1H),2.48~2.57(m,1H),3.55(d,J=5 Hz,1H),3.79(d,J=7 Hz,1H),4.25(AB q,JAB=8.4 Hz,δA-δB=32,2H),4.37~4.44(m,1H),4.80(dd,J=2.5和5 Hz,1H),4.94(bd,J=8.0 Hz,1H),5.67(d,J=7 Hz,1H),5.78(dd,J=2.5和8.8 Hz,1H),6.23(t,J=9 Hz,1H),6.27(s,1H),7.01(d,J=8.9 Hz,1H),7.33~7.54(m,10H),7.59~7.64(m,1H),7.22~7.75(m,2H),8.12~8.15(m,2H).13C-NMR δ:9.57(CH3),14.83(CH3),20.83(CH3),21.62(CH3),22.85(CH3),26.69(CH3),35.65(CH3),35.73(CH3),43.20(C),45.86(CH),55.05(CH),58.67(C),72.20(CH),72.41(CH),73.23(CH),75.00(CH),75.58(CH),76.58(CH2),79.10(C),81.21(C),84.42(CH),127.06(CH),128.38(CH),128.72(CH),129.04(CH),129.21(C),130.22(CH),131.37(CH),133.26(C),133.71(CH),138.03(C),141.98(C),167.04(C),170.17(C),171.22(C),172.73(C),203.62(C).与文献〔9〕值相符,即与红豆杉提取纯化的紫杉醇结构一致.半合成紫杉醇纯度测定依据HPLC法〔10〕,其纯度为99.6%.
, http://www.100md.com
1通讯联系人
参考文献
[1] Kingston DGI,Molinero AA,Rimoldi JM.The taxane diterpenoids.In:Herz W,Kirby GW,Moore RE,et al. eds,Progress in the Chemistry of Organic Natural Products,Vol.61,New York:Spiringer-Verlag,1993.31~60
[2] Holton RA,Somoza C,Kim HB, et al.First total synthesis of taxol.J Am Chem Soc,1994,116(14):1579~1602
[3] Nicolaou KC,Yang Z,Liu JJ,et al.Total synthesis of taxol.Nature,1994,367(17):630~632
, 百拇医药
[4] 阎家麒,刘虹,王九一,等.东北红豆杉原料紫杉醇的提取及测定.中国医药工业杂志,1994,25(10):433~436
[5] 阎家麒,范金城,王九一.紫杉醇提取纯化工艺.中国医药工业杂志,1996,27(12):531~534
[6] 陈冲,罗思齐.10-脱乙酰浆果杉亭-Ⅲ的新药源研究.中国医药工业杂志,1997,28(8):375~376
[7] Denis JN,Greene AE.A highly efficient practical approach to natural taxol. Am Chem Soc,1988,110(17):5917~5919
[8] 岳琴,方起程,梁晓天.紫杉醇的半合成.药学学报,1996,31(12):911~917
[9] Chmurny GN,Hilton BD,Brobst S,et al.1H-and 13C-NMR assignments for taxol,7-epitaxol and cephalomannine.J Nat Prod,1992,55(4):414~423
[10] 阎家麒,周海龙,周龙云,等.紫杉醇原料药纯度的RP-HPLC测定.中国医药工业杂志,1996,27(10):455~457
收稿日期:1998-10-24, http://www.100md.com(王九一1 阎家麒)