盐酸西替利嗪的合成
作者:李荣东 朱志宏 段立新
单位:李荣东(湖南省医药工业研究所,长沙 410014);朱志宏(湖南省医药工业研究所,长沙 410014);段立新(湖南省医药工业研究所,长沙 410014)
关键词:盐酸西替利嗪;抗变态反应;合成
摘 要摘 要:以氯苯和苯甲酰氯为起始原料,经付克反应、还原、氯化,再分别与哌嗪、氯乙醇、氯乙酸钠缩合,成盐制得盐酸西替利嗪,总收率为28.2%.
Synthesis of Cetirizine Hydrochloride
Li Rongdong Zhu Zhihong Duan Lixin
(Hunan Institute of Pharmaceutical Industry,Changsha 410014)
, http://www.100md.com
Abstract:Cetirizine hydrochloride was synthesized from chlorobenzene and benzoyl chloride by the Friedel-Crafts reaction,reduction,and chlorination followed by condensation with piperizine,2-chloroethanol and sodium chloroacetate in turn in 28.2%overall yield based on chlorobenzene.
Key words;cetirizine hydrochloride;anti-allergy;synthesis▲
盐酸西替利嗪(cetirizine hydrochloride)为第二代H1受体拮抗剂,为长效的选择性口服强效抗变态反应药.临床上广泛用于呼吸系统、皮肤和眼部过敏性疾病,并可以用于儿童.该品由比利时UCB公司研制,于1987年首次在比利时上市,1996年获FDA批准,现已在包括中国在内的几十个国家销售,湖南医药工业研究所于1995年开始研制本品,现已获得新药证书.
, 百拇医药
1 合成路线设计
盐酸西替利嗪为一外消旋产物,其合成路线主要有3条〔1~3〕,全部是以1-[(4-氯苯基)苯甲基]哌嗪(6)为起始原料.文献〔1〕以(6)与氯乙醇反应再与氯乙酸钠缩合.文献〔2〕以(6)与(2-氯乙氧基)乙酸甲酯反应,再水解.文献〔3〕以(6)与(2-氯乙氧基)乙腈反应再水解.文献〔2〕报道的路线两步反应收率太低,仅为10.6%.文献〔3〕路线由于中间体(2-氯乙氧基)乙腈的制备要用氰化亚铜,反应后过量的氰化亚铜难以处理,产品不好分馏,且纯度很低,因此选择了文献〔1〕的路线,并且对其进行了很大的改进.(6)与氯乙醇反应的产物(7)无需蒸馏,直接与氯乙酸钠反应,该反应过程中,文献采用分批加叔丁醇钾和氯乙酸钠的方法,操作过程由于叔丁醇钾有强吸湿性和强腐蚀性,且制备困难,操作极不方便.实验中改用一次性制备叔丁醇钾溶液的方法,生成的西替利嗪直接用盐酸成盐,用丁酮重结晶,三步反应的收率为49.8%(文献〔1〕收率:44.2%).1-[(4-氯苯基)苯甲基]哌嗪(6)的制备,采用自行设计的路线,以氯苯和苯甲酰氯为起始原料,经付克反应、还原、氯化再与无水哌嗪缩合制得目标化合物,并且对(5)与哌嗪缩合的工艺进行了很大改进,收率从文献〔4〕的47%上升到75.5%.目标化合物的合成路线见图1.
, http://www.100md.com
Fig.1 The synthetic route for cetirizine hydrochloride
该工艺原料全部国产化,操作简便,收率较文献有较大提高,质量可控制,适合工业化生产的要求.
2 实验部分
熔点采用毛细管法测定,温度未较正.元素分析仪为美国PE 2400型元素分析仪,红外光谱仪为Nicolet 510 P型.
2.1 4-氯双苯酮(3)的制备
向反应瓶中加入56.3 g(0.5 mol)氯苯(2)和73.4 g(0.55 mol)无水三氯化铝,升温至100℃,于100~110℃滴加70.3 g(0.5 mol)苯甲酰氯,滴完后继续反应6 h,然后缓缓倒入500 mL冰水中,静置,抽滤,水洗至中性,干燥得类白色固体(3)102.5 g,收率:94.7%,mp 74~76℃(文献〔5〕mp 75~76 ℃).
, 百拇医药
2.2 4-氯双苯甲醇(4)的制备
向反应瓶中加入60 g(1.5 mol)氢氧化钠,97.4 g(0.45 mol)4-氯双苯酮(3),300 mL 50%乙醇水溶液,加热至60℃,加入锌粉27 g(0.45 mol)于60~70℃反应1.5 h,再加27 g(0.45 mol)锌粉继续反应4 h,加热蒸除乙醇,冷却,加甲苯600 mL,抽滤,滤液分出甲苯层,母液用甲苯(200 mL×2)提取,无水硫酸钠干燥甲苯液,蒸干甲苯,得棕色固体(4)89.5 g,收率:91.0%.mp 59~61℃(文献〔6〕mp 60~61℃).
2.3 4-氯双苯氯甲烷(5)的制备
将87.4 g(0.4 mol)4-氯双苯甲醇(4),150 mL甲苯加入反应瓶中,滴加30 mL三氯氧磷,回流2 h,冷却,倒入冰水中,分出甲苯层,用饱和碳酸氢钠洗两次,水洗至中性,无水硫酸钠干燥,蒸干甲苯得棕色油状液体(5)82.5 g,收率:87.0%(直接用于下一步反应).
, http://www.100md.com
2.4 1-[(4-氯苯基)苯甲基]哌嗪(6)的制备
向反应瓶中加入86 g(1.0 mol)无水哌嗪,300 mL甲苯,加热至60℃,滴加82.5 g(0.348 mol)4-氯双苯氯甲烷(5),滴加完后,回流8 h,冷却,分出甲苯层,用50 mL浓盐酸稀释液洗甲苯二次,盐酸液用5 mol/L氢氧化钠中和至pH 10~11,再用甲苯(150 mL×2)提取,无水硫酸钠干燥,先蒸掉甲苯和少量哌嗪,然后减压蒸馏,收集240~245℃/2.0 kPa馏分(6)75.3 g,收率:75.5%(文献〔4〕190~192℃/240 Pa).
2.5 2-[4-[(4-氯苯基)苯甲基]-1-哌嗪基]乙醇(7)的制备
化合物(6)71.6 g(0.25 mol),甲苯150 mL,三乙胺60 mL,氯乙醇24.2 g(0.30 mol)加热回流反应6 h,再加入氯乙醇12.1 g(0.15 mol)继续回流16 h,冷却,过滤,滤液用大量热水洗涤,无水硫酸钠干燥,蒸干甲苯,得棕色油状物(7)72.8 g,收率:88.1%(直接用于下步反应).
, http://www.100md.com
2.6 2-[2-[4-[(4-氯苯基)苯甲基]-1-哌嗪基]乙氧基]-乙酸-盐酸盐(1)的合成
向反应瓶中加入500 mL叔丁醇,氮气保护下分批加金属钾17.55 g(0.45 mol),待金属钾完全反应后加入化合物(7)72.8 g(0.22 mol),于70~80 ℃搅拌1 h,然后加氯乙酸钠25.6 g(0.22 mol),回流反应2 h,再加氯乙酸钠12.8 g(0.11 mol),回流反应1.5 h,再加氯乙酸钠6.4 g(0.055 mol),继续回流反应8 h,蒸干叔丁醇,母液加水(200 mL)稀释,加浓盐酸调pH 8,用乙酸乙酯(100 mL×2)提取,再用浓盐酸调pH 5,二氯甲烷(150 mL×3)提取,无水硫酸钠干燥,蒸干二氯甲烷,残余物加浓盐酸30 mL和100 mL水组成的盐酸液,减压蒸除盐酸,蒸干后加丁酮300 mL回流1 h,析出白色固体,过滤,烘干,得白色固体(1)51.5 g,收率:56.5%.元素分析C21H25ClN2O3。2HCl,计算值(%):C 54.62,H 5.89,N 6.07;实测值(%):C 54.34,H 5.92,N 5.99.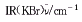 :3044(C―H),2949(C―H),2380(N―H),1741(C
:3044(C―H),2949(C―H),2380(N―H),1741(C O),1496(CC),1435(CN),1138(COC).■
O),1496(CC),1435(CN),1138(COC).■
, http://www.100md.com
参考文献:
[1]Cossement E,Gobert J,Bodson G.Process for the preparation of cetirizine and its dihydrochloride via etherification with haloacetates.Brit UK Pat Appl.GB 2225320.1990-05-30(CA 1990,113:191395s)
[2]Cossement E, Motte G, Bodson G, et al. Process for
preparation of cetirizine,its dihydrochloride and optical isomers via hydrolysis and corresponding nitriles.Brit UK Pat Appl.GB 2225321.1990-08-30(CA 1990,113:191396t)
, 百拇医药
[3]Baltes E,De Lannoy J,Rodriguez L.2-[4-(diphenylmethyl)-1-piperazinyl]acetic acids and their amides and pharmaceutical compositions.Eur Pat Appl.EP 58146.1982-08-18(CA 1983,98:34599r)
[4]Yung DK,Gilroy ML,Mahony DE.Synthesis and quantitative structure-activity relationships of antibacterial 1-(substituted benzhydryl-4-(5-nitro-2-furfurylideneamino)piperazines.J Pharm Sci,1978,67(7):900~905
[5]Staab HA,Jost E.Synthesis of ketones from inidazolides with Grignard reagents.Ann,1962,655:90~94(CA 1962,57:15091i)
[6]Roben HB, Boren EL. Catalysis with(-)- aluminum 2-
methybutoxide.J Am Chem Soc,1949,71(2):1399~1401
收稿日期:1999-08-03, http://www.100md.com
单位:李荣东(湖南省医药工业研究所,长沙 410014);朱志宏(湖南省医药工业研究所,长沙 410014);段立新(湖南省医药工业研究所,长沙 410014)
关键词:盐酸西替利嗪;抗变态反应;合成
摘 要摘 要:以氯苯和苯甲酰氯为起始原料,经付克反应、还原、氯化,再分别与哌嗪、氯乙醇、氯乙酸钠缩合,成盐制得盐酸西替利嗪,总收率为28.2%.
Synthesis of Cetirizine Hydrochloride
Li Rongdong Zhu Zhihong Duan Lixin
(Hunan Institute of Pharmaceutical Industry,Changsha 410014)
, http://www.100md.com
Abstract:Cetirizine hydrochloride was synthesized from chlorobenzene and benzoyl chloride by the Friedel-Crafts reaction,reduction,and chlorination followed by condensation with piperizine,2-chloroethanol and sodium chloroacetate in turn in 28.2%overall yield based on chlorobenzene.
Key words;cetirizine hydrochloride;anti-allergy;synthesis▲
盐酸西替利嗪(cetirizine hydrochloride)为第二代H1受体拮抗剂,为长效的选择性口服强效抗变态反应药.临床上广泛用于呼吸系统、皮肤和眼部过敏性疾病,并可以用于儿童.该品由比利时UCB公司研制,于1987年首次在比利时上市,1996年获FDA批准,现已在包括中国在内的几十个国家销售,湖南医药工业研究所于1995年开始研制本品,现已获得新药证书.
, 百拇医药
1 合成路线设计
盐酸西替利嗪为一外消旋产物,其合成路线主要有3条〔1~3〕,全部是以1-[(4-氯苯基)苯甲基]哌嗪(6)为起始原料.文献〔1〕以(6)与氯乙醇反应再与氯乙酸钠缩合.文献〔2〕以(6)与(2-氯乙氧基)乙酸甲酯反应,再水解.文献〔3〕以(6)与(2-氯乙氧基)乙腈反应再水解.文献〔2〕报道的路线两步反应收率太低,仅为10.6%.文献〔3〕路线由于中间体(2-氯乙氧基)乙腈的制备要用氰化亚铜,反应后过量的氰化亚铜难以处理,产品不好分馏,且纯度很低,因此选择了文献〔1〕的路线,并且对其进行了很大的改进.(6)与氯乙醇反应的产物(7)无需蒸馏,直接与氯乙酸钠反应,该反应过程中,文献采用分批加叔丁醇钾和氯乙酸钠的方法,操作过程由于叔丁醇钾有强吸湿性和强腐蚀性,且制备困难,操作极不方便.实验中改用一次性制备叔丁醇钾溶液的方法,生成的西替利嗪直接用盐酸成盐,用丁酮重结晶,三步反应的收率为49.8%(文献〔1〕收率:44.2%).1-[(4-氯苯基)苯甲基]哌嗪(6)的制备,采用自行设计的路线,以氯苯和苯甲酰氯为起始原料,经付克反应、还原、氯化再与无水哌嗪缩合制得目标化合物,并且对(5)与哌嗪缩合的工艺进行了很大改进,收率从文献〔4〕的47%上升到75.5%.目标化合物的合成路线见图1.
, http://www.100md.com
Fig.1 The synthetic route for cetirizine hydrochloride
该工艺原料全部国产化,操作简便,收率较文献有较大提高,质量可控制,适合工业化生产的要求.
2 实验部分
熔点采用毛细管法测定,温度未较正.元素分析仪为美国PE 2400型元素分析仪,红外光谱仪为Nicolet 510 P型.
2.1 4-氯双苯酮(3)的制备
向反应瓶中加入56.3 g(0.5 mol)氯苯(2)和73.4 g(0.55 mol)无水三氯化铝,升温至100℃,于100~110℃滴加70.3 g(0.5 mol)苯甲酰氯,滴完后继续反应6 h,然后缓缓倒入500 mL冰水中,静置,抽滤,水洗至中性,干燥得类白色固体(3)102.5 g,收率:94.7%,mp 74~76℃(文献〔5〕mp 75~76 ℃).
, 百拇医药
2.2 4-氯双苯甲醇(4)的制备
向反应瓶中加入60 g(1.5 mol)氢氧化钠,97.4 g(0.45 mol)4-氯双苯酮(3),300 mL 50%乙醇水溶液,加热至60℃,加入锌粉27 g(0.45 mol)于60~70℃反应1.5 h,再加27 g(0.45 mol)锌粉继续反应4 h,加热蒸除乙醇,冷却,加甲苯600 mL,抽滤,滤液分出甲苯层,母液用甲苯(200 mL×2)提取,无水硫酸钠干燥甲苯液,蒸干甲苯,得棕色固体(4)89.5 g,收率:91.0%.mp 59~61℃(文献〔6〕mp 60~61℃).
2.3 4-氯双苯氯甲烷(5)的制备
将87.4 g(0.4 mol)4-氯双苯甲醇(4),150 mL甲苯加入反应瓶中,滴加30 mL三氯氧磷,回流2 h,冷却,倒入冰水中,分出甲苯层,用饱和碳酸氢钠洗两次,水洗至中性,无水硫酸钠干燥,蒸干甲苯得棕色油状液体(5)82.5 g,收率:87.0%(直接用于下一步反应).
, http://www.100md.com
2.4 1-[(4-氯苯基)苯甲基]哌嗪(6)的制备
向反应瓶中加入86 g(1.0 mol)无水哌嗪,300 mL甲苯,加热至60℃,滴加82.5 g(0.348 mol)4-氯双苯氯甲烷(5),滴加完后,回流8 h,冷却,分出甲苯层,用50 mL浓盐酸稀释液洗甲苯二次,盐酸液用5 mol/L氢氧化钠中和至pH 10~11,再用甲苯(150 mL×2)提取,无水硫酸钠干燥,先蒸掉甲苯和少量哌嗪,然后减压蒸馏,收集240~245℃/2.0 kPa馏分(6)75.3 g,收率:75.5%(文献〔4〕190~192℃/240 Pa).
2.5 2-[4-[(4-氯苯基)苯甲基]-1-哌嗪基]乙醇(7)的制备
化合物(6)71.6 g(0.25 mol),甲苯150 mL,三乙胺60 mL,氯乙醇24.2 g(0.30 mol)加热回流反应6 h,再加入氯乙醇12.1 g(0.15 mol)继续回流16 h,冷却,过滤,滤液用大量热水洗涤,无水硫酸钠干燥,蒸干甲苯,得棕色油状物(7)72.8 g,收率:88.1%(直接用于下步反应).
, http://www.100md.com
2.6 2-[2-[4-[(4-氯苯基)苯甲基]-1-哌嗪基]乙氧基]-乙酸-盐酸盐(1)的合成
向反应瓶中加入500 mL叔丁醇,氮气保护下分批加金属钾17.55 g(0.45 mol),待金属钾完全反应后加入化合物(7)72.8 g(0.22 mol),于70~80 ℃搅拌1 h,然后加氯乙酸钠25.6 g(0.22 mol),回流反应2 h,再加氯乙酸钠12.8 g(0.11 mol),回流反应1.5 h,再加氯乙酸钠6.4 g(0.055 mol),继续回流反应8 h,蒸干叔丁醇,母液加水(200 mL)稀释,加浓盐酸调pH 8,用乙酸乙酯(100 mL×2)提取,再用浓盐酸调pH 5,二氯甲烷(150 mL×3)提取,无水硫酸钠干燥,蒸干二氯甲烷,残余物加浓盐酸30 mL和100 mL水组成的盐酸液,减压蒸除盐酸,蒸干后加丁酮300 mL回流1 h,析出白色固体,过滤,烘干,得白色固体(1)51.5 g,收率:56.5%.元素分析C21H25ClN2O3。2HCl,计算值(%):C 54.62,H 5.89,N 6.07;实测值(%):C 54.34,H 5.92,N 5.99.
, http://www.100md.com
参考文献:
[1]Cossement E,Gobert J,Bodson G.Process for the preparation of cetirizine and its dihydrochloride via etherification with haloacetates.Brit UK Pat Appl.GB 2225320.1990-05-30(CA 1990,113:191395s)
[2]Cossement E, Motte G, Bodson G, et al. Process for
preparation of cetirizine,its dihydrochloride and optical isomers via hydrolysis and corresponding nitriles.Brit UK Pat Appl.GB 2225321.1990-08-30(CA 1990,113:191396t)
, 百拇医药
[3]Baltes E,De Lannoy J,Rodriguez L.2-[4-(diphenylmethyl)-1-piperazinyl]acetic acids and their amides and pharmaceutical compositions.Eur Pat Appl.EP 58146.1982-08-18(CA 1983,98:34599r)
[4]Yung DK,Gilroy ML,Mahony DE.Synthesis and quantitative structure-activity relationships of antibacterial 1-(substituted benzhydryl-4-(5-nitro-2-furfurylideneamino)piperazines.J Pharm Sci,1978,67(7):900~905
[5]Staab HA,Jost E.Synthesis of ketones from inidazolides with Grignard reagents.Ann,1962,655:90~94(CA 1962,57:15091i)
[6]Roben HB, Boren EL. Catalysis with(-)- aluminum 2-
methybutoxide.J Am Chem Soc,1949,71(2):1399~1401
收稿日期:1999-08-03, http://www.100md.com