甾体17 α ―羟化酶/C17,20- 裂解酶(P45017 α)抑制剂的三维定量构效关系研究
http://www.100md.com
中国药学会
 |
 |
 |
 |
 |
苗 及 凌仰之 朱 娜 雷小平
北京大学药学院药物化学系 北京100083
摘 要 目的:建立P45017(抑制剂的三维定量构效关系,为设计新的更有效的抑制剂提供理论依据。方法和结果:利用比较分子力场方法,建立了P45017(抑制剂的三维定量构效关系模型。交叉验证回归系数R2CV、非交叉验证回归系数和标准偏差SEE分别为0.533,0.776,0.271。说明系列化合物分子周围立体场和静电场的分布与生物活性间有良好的相关性。利用该模型对本室合成的三个化合物进行活性预测,结果与实测值相符。结论:所得模型支持了假设的抑制剂作用机理和作用模型。所得勘MFA模型具有一定的预测能力,可用来指导设计新的P45017α抑制剂。
, 百拇医药 关键词 P45017α抑制剂 比较分子场分析法(CoMFA) 三维定量构效关系模型
17α―羟化酶/Cl7,20裂解酶是位于睾丸和肾上腺的细胞色素酶。它是雄激素生物合成途径中的关键酶。功能是将2l碳的生物前体孕烯醇酮和孕酮分别在两步中去C20―乙酰侧链和17位羟化,转化成19碳的去氢表雄酮和雄烯二酮。最后由位于前列腺中的5α―还原酶将睾酮转化成二氢睾酮。已经证明体内的一些疾病是由体内过高的雄激素导致的,如前列腺良性增生,前列肠癌等。临床上已证明降低体内的雄激素水平是治疗这些疾病的有效方法。选择性高、作用强的P45017α酶抑制剂可以通过抑制雄激素生物合成中较初始环节,降低体内雄激素水平,在临床上用于治疗前列腺疾病。已报道的P45017α。抑制剂主要包括国体类和非甾体类,而非团体类抑制剂大都不具有好的选择性。为了进一步阐明P45017α抑制剂的定量构效关系,建立具有提示高活性化合物信息及预测能力的构效关系模型,本文利用比较分子力场方法(CoMFA),对甾体类抑制剂进行了三维定量构效关系研究。
, 百拇医药
实验方法
所有工作都是在Tripos公司SiliconGraphicsO2计算机工作站上完成的。所用计算程序为Tripos公司的Sybyl6.4分子设计软件包。计算过程所涉及的参数除非特别指明,均为缺省值。
1 化合物的选择 P450l7α(CYPl7)是一种结构尚未知的细胞色素酶。其抑制剂的筛选有多种方法。因此对化合物的活性数据进行了严格考察。本文选择了31个团体抑制剂进行CoMFA研究。它们的活性数据源于同一个实验室。每次测试中,对照样品活性值相同,认为有可比性。所选取样品是以酮康唑为标准品,其IC50值为78nM。比较分子力场分析采用IC50的负对数形式―10gIC50。所选用的化合物结构及活性见表l。
Tabl The structures and activities of 31 compounds used in CoMFA.
, 百拇医药
No.
Basic Structure
R1
R2
R3
R
IC50(nM)
-1og1/c
1
B
OH
H
, 百拇医药
H
(a)
66
4,18
2
B
OH
α-O-
(a)
430
3.37
3
B
OH
, http://www.100md.com
C=C
(a)
24
4.62
4
B
OAc
C=C
(a)
75
4.12
5
B
OAc
, 百拇医药
H
H
(a)
199
3.70
6
B
OAc
H
H
(a) X=Ac
100
4.00
, 百拇医药 7
A
O
H
H
(a)
58
4.24
8
A
O
OH
H
(a)
, 百拇医药
1200
2.92
9
A
O
C=C
(a)
50
4.30
10
B
OH
C=C
(b)
, http://www.100md.com
21
4.68
11
B
OH
C=C
(c) X,Y=N
42
4.38
12
B
OH
C=C
, http://www.100md.com
(c) X=O,Y=N
108
3.97
13
B
OH
H
H
(c) X=N,Y=O
1113
2.95
14
A
, 百拇医药
O
C=C
(c) X=N,Y=N
59
4.23
15
A
O
C=C
(c) X=O,Y=O
39
4.41
16
, 百拇医药
B
OH
C=C
(d) X=N-OH
73
4.14
17
B
OAc
C=C
(d) X=N-OH
67
4.17
, 百拇医药
18
B
OAc
C=C
(d) X=OAc
51
4.29
19
B
OH
(e) X=H,Y=Me
339
3.47
, 百拇医药
20
B
OH
(e) X=Me,Y=H
372
3.43
21
B
OAc
α-N-
(d) X=O
162
3.79
, http://www.100md.com
22
A
O
H
H
(d) X=N―OH
43
4.37
23
A
O
H
H
(d) X=N―OAc
, http://www.100md.com
25
4.60
24
A
NOH(E)
C=C
(d) X=N―OH
196
3.7l
25
A
NOH(Z)
C=C
, 百拇医药
(d)
(d) X=N―OH
75
4.12
26
A
O
C=C
(f) X=N,Y=N,Z=C
90
4.72
27
B
, 百拇医药
OH
C=C
(f) X=N,Y=C,Z=N
8
4.04
28
B
OH
C=C
(f) X=C,Y=N,Z=C
13
5.10
29
, http://www.100md.com
B
OH
C=C
(f) X=N,Y=N,Z=C
55
4.88
30
A
O
C=C
(f) X=N,Y=C,Z=N
7
4.26
, 百拇医药
31
A
O
C=C
(f) X=N,Y=N,Z=C
19
5.15
2 活性构象确定 由于酶结构未知,所以采用分子的最低能量构象作为最可能的药效构象。以活性最好的化合物31作为模板分子。其起始构象是在Sybyl6.4的“Build/Edit”模块下建立,用分子力学程序Minimize进行能量优化。优化过程中采用Powell能量梯度法,用Tripos力场,能量收敛限定为0.001kcal/mol-l,优化次数为1000。分子载荷为Gasteiger―Huchel型电荷。其它化合物以3l为模板,变换成相应结构,也以Minimize程序以同样条件能量优化后,作为最低能量构象。
, http://www.100md.com
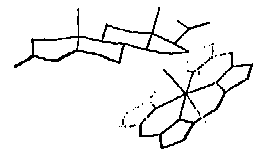
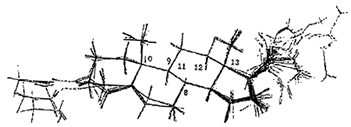
3 分子叠合 由于抑制剂同属甾体类,它们与酶结合时部位应一致。而它们之间除17位取代不同外,仅是A环和D环有差别。因此以活性最好的化合物31为模板,以B环和C环上的C8,C9,C10,C11,C12,C13,为叠合点进行叠合。叠合图见图1
Fig 1:Stereoview of superimposition of 31 P45017αinhinbitors used in CoMFA.
4 CoMFA分析 CoMFA分析由Sybyl6.4中的QSAR模块来完成。分析力场包括立体场和静电场。在0-50kal/mol-1范围内,经多次摸索,将立体场和静电场的能量域值分别设为15kcal/mol-1和15kcal/mol-1。先以交叉验证(cross-validation)确定最佳主成分数,再用非交叉验证(non-validation)建立最终的比较分子力场模型。在最小二乘法(Partial Least Square,PLS)分析中,column filtering设为4kcal/mol-1。
, 百拇医药
结果与讨论
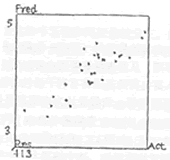
1 CoMPA模型 比较分子力场分析所得勘MFA模型的统计学参数为:交叉验证相关系数R2cv=0.533,最佳主成分数为3,由最佳主成分数建立的CoMFA模型的传统相关系数R2=0.799,F=31.201,标准方差SEE(standard error of estimate)=0.271。该模型对31个化合物的活性预测值与实测值的相关性如图2。
Fig 2:Predicted vs actual biological activities of 31 inhibitiors from CoMFA model
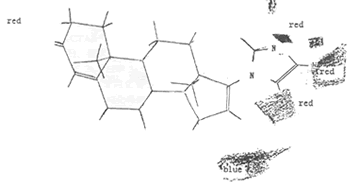
2.CoMFA 系数等势图 CoMFA模型系数等势图见图3(a)和3(b).其中参照分子化合物31。图3(a)中绿色和黄色区域代表立体场对化合物活性的影响,绿色区域表示在该区域附近存在体积大的基团将由于增加的立体场面有利于活性的提高,黄色区域则表示该区域不宜于引入位阴大的基团,否则会降低化合物的活性。
, 百拇医药
图3(b)中红色和兰色区域代表静电场对活性的影响:红色区域表示引入负电性基团有利于提高活生,兰色区域则表明引入正电性基团有利于活性提高。
Fig 3(a):View of the CoMFA steric field graph
Fig 3(b):View of the CoMFA electrostatic field graph
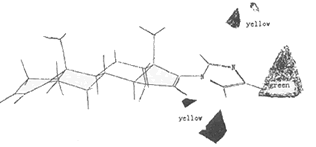
3 关于模型的讨论 在立体场等势图中可以看到,在甾体骨架的(面有一块黄色区域,说明在此处存在有位阻大的基团将不利于抑酶作用;从静电场等势图中可以看出在化合物31的杂环附近有多处红色区域,说明在这些区域引入富电荷的基团将有利于活性的提高。这与文献中假设的竞争性P450l7α抑制剂的作用机理和作用模型相符。该假设认为在距离甾体D环适当位置引入具有孤对电子的原子,如N等,将可能与P450l7α的血红素辅基中的Fe原子结合,而抑制酶的活性。假设的作用模式中是血红素位于甾环的α面(如图4),我们得到的3DCoMFA模型更进一步说明了,在甾环D环存在富电荷的原子有利于抑酶活性,这支持了上述作用机理。α面位阻大基团不利于抑酶活性,这是与假设的作用模型一致,因为在此处有位阻大的基团,会阻碍杂原子与Fe作用,而不利于抑酶活性。
, http://www.100md.com
Fig 4:Stereoplot of the substrate heme-complex for the P45017α
利用该模型对本室合成的3个化合物进行了活性预测。预测结果是以3―羟基―17―(2’―噻唑)雄甾―5,16二烯中为母体的系列中4’位苯基取代衍生物的活性(4.76)大于4,位氢取代(4.61)大于4,位甲基取代(4.47)。由于这三个化合物活性的测定与勘MFA模型建立所用的化合物活性的测定是在不同条件下进行,对照品采用VIN85而非酮康唑,因而活性数据没有直接的可比性。但实测的抑制率的顺序与预测顺序相同,是4’位苯基取代大于4’位氢取代大于4’位甲基取代。说明该模型对于抑制剂的电性、立体要求有较为准确的描述。
本文摘自2000北京地区药学学术年会《大会论文集》, 百拇医药