1-[(2-羟乙氧基)甲基]-6-(苯硫基)胸腺嘧啶类化合物的合成及构效关系研究进展
作者:张铭龙 马灵台 张礼和1
单位:北京医科大学药学院国家天然仿生药物重点实验室,北京 100083
关键词:1-[(2-羟乙氧基)甲基]-6-(苯硫基)胸腺嘧啶;抗病毒;艾滋病病毒逆转录酶;构效关系
中国药物化学杂志990117 摘 要 介绍了一类新型抗病毒化合物1-[(2-羟乙氧基)甲基]-6-(苯硫基)胸腺嘧啶的结构特点、作用机理及构效关系研究.并比较了它与其它抗病毒药物对艾滋病病毒作用的机理、病毒的耐药机理以及联合用药的研究情况.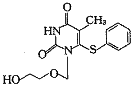
1-[(2-羟乙氧基)甲基]-6-(苯硫基)胸腺嘧啶(HEPT)是
一个开链核苷酸,是胸腺嘧啶的1位氮上连结―CH2OCH2CH2OH基,6位碳上联结苯硫基结合而成.由于它仅拥有核苷类化合物的碱基,不具有核苷类化合物的糖环,所以把它列为非核苷类化合物.它是继无环鸟苷(acyclovir)后的又一个含有核苷碱基而不含糖环的具有抗病毒活性的非核苷类化合物,因而受到广泛重视〔1~3〕.近年世界各国的科学家以HEPT骨架为基础进行结构改造,以期寻找到高效低毒的新一类抗病毒药物.
, http://www.100md.com
由于病毒性疾病已成为对人类伤害最大的疾病之一,尤其是艾滋病的蔓延,引起了广泛的关注,到目前为止,已被美国食品药品管理局(FDA)批准上市的抗艾滋病药物有齐多夫定(AZT)、地丹诺辛(DDI)、扎西它宾(DDC)、司他夫定(D4T)、拉米夫定(3Tc)、沙奎那韦(saquinavir)、利托那韦(ritonavir)和英地那韦(indinavir)等,除后3者外,它们的结构都属于核苷类化合物.在体内干扰病毒正常的RNA或DNA复制,从而抑制病毒的繁殖,而产生抗病毒作用.但它同时也干扰了人体正常的DNA复制,从而产生了强烈的毒副作用.
为降低药物的毒性,提高疗效,科学家开始将目光转向对非核苷类化合物的研究,它已成为抗病毒药物研究的重点.目前已进入艾滋病临床研究阶段的化合物主要有:bis(heteroaryl)piperazine双杂芳哌嗪类(代表化合物U 87261)、TIBO类、TSAO类(代表化合物TSAO-T)、nevirapine双吡啶类(代表化合物BI-GR-587)、2-pyridione吡啶酮类(代表化合物L-697,661),以及无环鸟苷等〔1〕.1 HEPT类化合物的生物活性
, 百拇医药
由于目前已进入艾滋病临床研究阶段的化合物大多是作用于病毒的逆转录酶,抑制病毒的RNA链通过逆转录酶整合到宿主细胞的DNA上,而起到抑制病毒繁殖的作用.这些化合物虽然降低了毒副作用,但它们用于临床不久就相继产生了病毒的耐药株,这就极大地限制了这些化合物在临床上的应用.尽管如此,研究发现,这些化合物所引起的HIV变异株却各不相同.如双杂芳哌嗪类化合物表现为病毒逆转录酶蛋白的第100位氨基酸发生变化产生抗药性,而TIBO类化合物表现为病毒的第103位氨基酸发生变化产生抗药性,TSAO类化合物又表现为病毒的第138位氨基酸发生变化产生抗药性,双吡啶类化合物表现为病毒的第106位氨基酸发生变化而抗药,吡啶酮类化合物的抗药株病毒表现为在氨基酸第181位发生变化〔1〕.而HEPT类化合物的耐药株则主要表现为100位的亮氨酸变异为异亮氨酸,103位的赖氨酸变异为天门冬氨酸,106位的缬氨酸变异为丙氨酸,138位的谷氨酸变异为赖氨酸,181位的酪氨酸变异为半胱氨酸,以及188位的酪氨酸变异为组氨酸〔2〕.经过广泛的构效关系研究、合成、筛选,发现一些HEPT类化合物具有较强的HIV病毒抑制作用,而且毒副作用小.由于其它各类抗HIV药物使病毒产生的耐药株的变异各不相同,利用这一性质可以对各类抗HIV药物产生的耐药株进行交叉抑制.因此HEPT类化合物具有广泛的抗耐药作用〔1〕.进一步研究表明,HEPT仅对HIV-1型逆转录酶有抑制作用,而对HIV-2型逆转录酶则没有作用〔1〕.
, 百拇医药
尽管HEPT作为非核苷类药物具有低毒高效的作用,但是临床长期大剂量使用仍然会产生湿疹、转氨酶升高等副作用.因此为增加药效,减少副作用,避免HIV产生抗药性,将HEPT分别与临床上常用的抗艾滋病药物如:AZT、DDI、DDC和碳环鸟苷等进行联合用药,一方面通过不同化合物的不同抑制作用,对其它化合物引起的变异病毒进行抑制,另一方面利用它们的协同作用而产生强的对病毒抑制作用〔3〕.例如:R.W.Buckheit证实了HEPT不仅对多种核苷及非核苷类化合物的耐药株有抑制作用,而且考查了它与其它化合物的配伍作用.数据表明,HEPT与Calanolide A的配伍效果为最佳,它们几乎对实验所涉及到的所有耐药株都具有抑制作用〔3〕.
2 HEPT类化合物的结构修饰
有关HEPT类化合物的合成及结构改造已有多条合成路线.
2.1 对6位取代基团的修饰改造路线〔4~7〕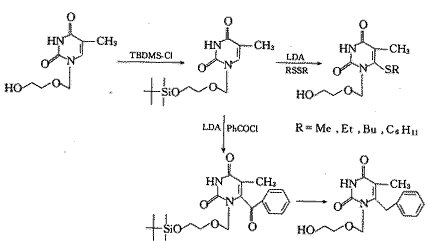
, 百拇医药
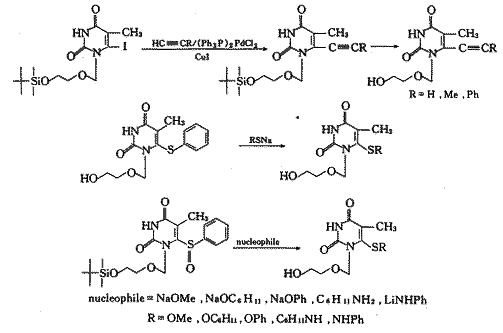
2.2 对苯环的修饰改造〔8,9〕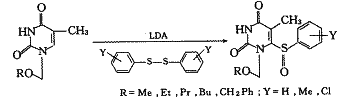
2.3 对5位取代基团的修饰改造〔5,9〕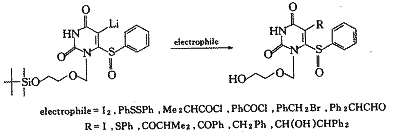
2.4 对1位侧链的修饰〔8,10〕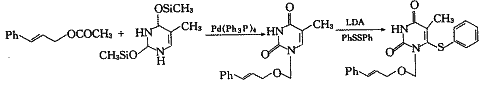 3 HEPT类化合物的构效关系
3 HEPT类化合物的构效关系
3.1 通过对HIV-1逆转录酶晶体的三维结构分析进行构效关系研究
通过对HEPT结构改造、研究,A.L.Hopkins对HIV-1逆转录酶晶体的三维结构进行了分析,通过对HEPT及其衍生物与HIV-1逆转录酶作用进行受力、构象及电性作用研究,计算机辅助设计、计算、分析得到如下HEPT构效关系规律〔11〕:1)HEPT类化合物的抗HIV作用主要是它们的6位芳环干扰HIV-1逆转录酶的181位酪氨酸的构象而起作用,干扰越强,则作用越强.2)连接胸腺嘧啶环与苯环结构的硫并不是必需基团,它可以被其它基团如亚甲基等取代.3)HEPT的5位取代基可以干扰HIV-1逆转录酶的181位酪氨酸的构象,如被大体积基团如异丙基取代时,则明显改变181位酪氨酸的构象,而加强6位芳环的干扰作用.4)HEPT与HIV-1逆转录酶的作用区域主要为疏水亲脂环境,对HEPT进行亲脂性改造将会有助于两者的结合.如将HEPT的1位末端羟基连接亲脂基团,将有利于化合物与酶的结合,提高活性.
, 百拇医药
3.2 通过将HIV-1逆转录酶抑制剂与HEPT连接进行构效关系研究
为了进一步研究联合用药的有效性、发展新的抗HIV品种,S.Velazquez将对艾滋病病毒有抑制作用的化合物如:AZT、TSAO等分别用不同长度的碳链与HEPT连接起来,进行药理实验.发现这种连接的产物虽然仍然具有抗HIV-1病毒活性,但其作用已不如连接前两个母体化合物各自的活性,而且对HIV-2无效〔12〕.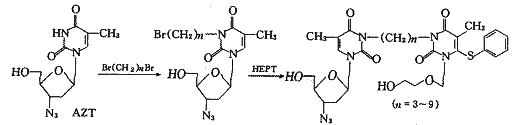
3.3 通过对HEPT的电性、亲脂性、空间结构及构象分析进行构效关系研究
M.H.Nguyen等在研究HEPT类化合物构效关系时发现,HEPT的5位乙基或异丙基取代物都要比甲基取代物活性高,如:5-乙基HEPT对HIV-1的EC50为0.1 μmol/L,而5-甲基HEPT的EC50仅为2.6 μmol/L〔3〕,而除去侧链末端亲水的羟基也可以提高化合物的活性〔4〕.J.Renault等认为体积较大的乙基由于空间位阻效应,阻挡了苯环的旋转,而使化合物的活性提高.由此推断将苯环锁住的平面结构可使苯环与胸腺嘧啶环形成大π键共轭体系,可能对提高化合物活性有利.另外经X光衍射晶体结构分析及计算机图形分析发现,HEPT与HIV逆转录酶的作用区主要为亲脂环境,因此设计了一系列五元及六元环结构的化合物将胸腺嘧啶环与苯环锁住,交叉地也把HEPT末端亲水的羟基或用亲脂性的苯环取代,或再增加一个亲水羟基,遗憾地是所合成的化合物中没有一个具有抗HIV活性〔4〕.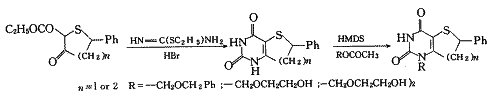
, 百拇医药
但是,H.Maruenda等受TIBO和双吡啶类化合物七元环结构的启发,用七元环把胸腺嘧啶环与苯环锁住,并将1位侧链末端亲水的羟基乙醚化以增加化合物的脂溶性,则得到了高活性的衍生物HEPT-25,它的EC50达到0.64 μmol/L〔14〕.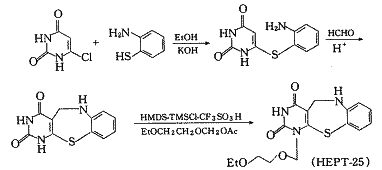
通过合成、药理筛选及构效关系分析,对HEPT类化合物的研究有了新的进展,已有更新、更有效的化合物进入了进一步实验研究,如化合物HEPT-33的EC50达到0.0042 μmol/L,现已进入临床实验阶段〔5〕.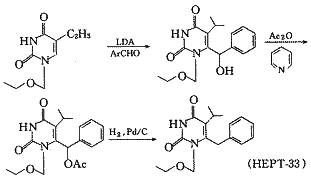
有关HEPT类化合物的合成及生物活性研究工作刚刚起步,方兴未艾.希望在不久的将来,能够有HEPT类的抗病毒新药问世.
, http://www.100md.com
1通讯联系人
参考文献
[1] Balzarini J,Karlsson A,Clercq ED.Human immunodeficiency virus type 1 drug-resistance patterns with different 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine derivative.Molecular Pharmacology,1993,44:694~701
[2] Balzarini J,Baba M,Clercq ED.Differential activities of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine derivatives against different human immunodeficiency virus type 1 mutant strains.Antimicronial Agents and Chemotherapy,1995,39(4):998~1002
, http://www.100md.com
[3] Robert W,Buckheit JR,Valerie FB,et al.Resistance to 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine derivatives is generated by mutations at multiple sites in the HIV-reverse transcriptase.Virology,1995,210:186~193
[4] Renault J,Laduree D,Robba M.Synthesis and antiviral study of dihydrothieno and thianopyrimidine diones acyclic nucleosides as potential anti-HIV agents.Nucleosides & Nucleotides,1994,13(5):1135~1145
[5] Tanaka H,Takashima H,Ubasawa M,et al.Synthesis and antiviral activity of 6-benzyl analogs of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine(HEPT)as potential and selsctive anti-HIV agents.J Med Chem,1995,38:2860~2865
, 百拇医药
[6] Miyasaka T,Tanaka H,Baba M,et al.A novel lead for specific anti-HIV-1 agent:1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine.J Med Chem,1989,32:2507~2509
[7] Tanaka H,Baba M,Hayakawa H,et al.A new class of HIV-1-specific 6-substituted acyclouridine derivatives:synthesis and anti-HIV-1 activity of 5-or 6-substituted analogues of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine(HEPT).J Med Chem,1991,34:349~357
[8] Pontikis R,Monneret C.Synthesis of deoxy analogs of HEPT involving a palladium(o)catalyzed coupling.Tetrahedron Lett,1994,35(25):4351~4354
, http://www.100md.com
[9] Tanaka H,Takashima H,Ubasawa M,et al.Synthesis and antiviral activity of deoxy analogs of 1-〔(2-hydroxyethoxy)methyl〕-6-(phenylthio)thymine(HEPT)as potential and selective anti-HIV agents.J Med Chem,1992,35:4713~4719
[10] Tanaka H,Baba M,Hayakawa H,et al.Lithiation of uracilnucleosides and its application to the synthesis of a new class of anti-HIV-1 acyclonucleosides.Nucleosides & Nucleotides,1991,10(1~3):379~400
[11] Hopkins AL,Ran J,Esnouf RM,et al.Complexes of HIV-1 reverse transcriptase inhibitors of the HEPT series reveal conformational changes relevant to the design of potent non-nucleoside inhibitors.J Med Chem,1996,39:1589~1600
, 百拇医药
[12] Velazqjez S,Alvarez R,San-Felix A,et al.Synthesis and anti-HIV activity of [AZT]-[TSAOT]and [AZT]-[HEPT]dimers as potential multifunctional inhibitors of HIV-1 reverse transcriptase.J Med Chem,1995,38:1641~1649
[13] Nguyen MH,Schinazi RF,Shi C.et al.Resistance of human immunodeficiency virus type 1 to acyclic 6-phenylsenenyl-and phenylthiopyrimidines.Antimicronial Agents and Chemotherapy,1994,38(10):2409~2414
[14] Maruenda H,Johnson F.Design and synthesis of novel inhibitors of HIV-1 reverse transcriptase.J Med Chem,1995,38(12):2145~2151
收稿日期:1998-10-19, 百拇医药(张铭龙 马灵台 张礼和1)
单位:北京医科大学药学院国家天然仿生药物重点实验室,北京 100083
关键词:1-[(2-羟乙氧基)甲基]-6-(苯硫基)胸腺嘧啶;抗病毒;艾滋病病毒逆转录酶;构效关系
中国药物化学杂志990117 摘 要 介绍了一类新型抗病毒化合物1-[(2-羟乙氧基)甲基]-6-(苯硫基)胸腺嘧啶的结构特点、作用机理及构效关系研究.并比较了它与其它抗病毒药物对艾滋病病毒作用的机理、病毒的耐药机理以及联合用药的研究情况.
1-[(2-羟乙氧基)甲基]-6-(苯硫基)胸腺嘧啶(HEPT)是
一个开链核苷酸,是胸腺嘧啶的1位氮上连结―CH2OCH2CH2OH基,6位碳上联结苯硫基结合而成.由于它仅拥有核苷类化合物的碱基,不具有核苷类化合物的糖环,所以把它列为非核苷类化合物.它是继无环鸟苷(acyclovir)后的又一个含有核苷碱基而不含糖环的具有抗病毒活性的非核苷类化合物,因而受到广泛重视〔1~3〕.近年世界各国的科学家以HEPT骨架为基础进行结构改造,以期寻找到高效低毒的新一类抗病毒药物.
, http://www.100md.com
由于病毒性疾病已成为对人类伤害最大的疾病之一,尤其是艾滋病的蔓延,引起了广泛的关注,到目前为止,已被美国食品药品管理局(FDA)批准上市的抗艾滋病药物有齐多夫定(AZT)、地丹诺辛(DDI)、扎西它宾(DDC)、司他夫定(D4T)、拉米夫定(3Tc)、沙奎那韦(saquinavir)、利托那韦(ritonavir)和英地那韦(indinavir)等,除后3者外,它们的结构都属于核苷类化合物.在体内干扰病毒正常的RNA或DNA复制,从而抑制病毒的繁殖,而产生抗病毒作用.但它同时也干扰了人体正常的DNA复制,从而产生了强烈的毒副作用.
为降低药物的毒性,提高疗效,科学家开始将目光转向对非核苷类化合物的研究,它已成为抗病毒药物研究的重点.目前已进入艾滋病临床研究阶段的化合物主要有:bis(heteroaryl)piperazine双杂芳哌嗪类(代表化合物U 87261)、TIBO类、TSAO类(代表化合物TSAO-T)、nevirapine双吡啶类(代表化合物BI-GR-587)、2-pyridione吡啶酮类(代表化合物L-697,661),以及无环鸟苷等〔1〕.1 HEPT类化合物的生物活性
, 百拇医药
由于目前已进入艾滋病临床研究阶段的化合物大多是作用于病毒的逆转录酶,抑制病毒的RNA链通过逆转录酶整合到宿主细胞的DNA上,而起到抑制病毒繁殖的作用.这些化合物虽然降低了毒副作用,但它们用于临床不久就相继产生了病毒的耐药株,这就极大地限制了这些化合物在临床上的应用.尽管如此,研究发现,这些化合物所引起的HIV变异株却各不相同.如双杂芳哌嗪类化合物表现为病毒逆转录酶蛋白的第100位氨基酸发生变化产生抗药性,而TIBO类化合物表现为病毒的第103位氨基酸发生变化产生抗药性,TSAO类化合物又表现为病毒的第138位氨基酸发生变化产生抗药性,双吡啶类化合物表现为病毒的第106位氨基酸发生变化而抗药,吡啶酮类化合物的抗药株病毒表现为在氨基酸第181位发生变化〔1〕.而HEPT类化合物的耐药株则主要表现为100位的亮氨酸变异为异亮氨酸,103位的赖氨酸变异为天门冬氨酸,106位的缬氨酸变异为丙氨酸,138位的谷氨酸变异为赖氨酸,181位的酪氨酸变异为半胱氨酸,以及188位的酪氨酸变异为组氨酸〔2〕.经过广泛的构效关系研究、合成、筛选,发现一些HEPT类化合物具有较强的HIV病毒抑制作用,而且毒副作用小.由于其它各类抗HIV药物使病毒产生的耐药株的变异各不相同,利用这一性质可以对各类抗HIV药物产生的耐药株进行交叉抑制.因此HEPT类化合物具有广泛的抗耐药作用〔1〕.进一步研究表明,HEPT仅对HIV-1型逆转录酶有抑制作用,而对HIV-2型逆转录酶则没有作用〔1〕.
, 百拇医药
尽管HEPT作为非核苷类药物具有低毒高效的作用,但是临床长期大剂量使用仍然会产生湿疹、转氨酶升高等副作用.因此为增加药效,减少副作用,避免HIV产生抗药性,将HEPT分别与临床上常用的抗艾滋病药物如:AZT、DDI、DDC和碳环鸟苷等进行联合用药,一方面通过不同化合物的不同抑制作用,对其它化合物引起的变异病毒进行抑制,另一方面利用它们的协同作用而产生强的对病毒抑制作用〔3〕.例如:R.W.Buckheit证实了HEPT不仅对多种核苷及非核苷类化合物的耐药株有抑制作用,而且考查了它与其它化合物的配伍作用.数据表明,HEPT与Calanolide A的配伍效果为最佳,它们几乎对实验所涉及到的所有耐药株都具有抑制作用〔3〕.
2 HEPT类化合物的结构修饰
有关HEPT类化合物的合成及结构改造已有多条合成路线.
2.1 对6位取代基团的修饰改造路线〔4~7〕
, 百拇医药
2.2 对苯环的修饰改造〔8,9〕
2.3 对5位取代基团的修饰改造〔5,9〕
2.4 对1位侧链的修饰〔8,10〕
3 HEPT类化合物的构效关系3.1 通过对HIV-1逆转录酶晶体的三维结构分析进行构效关系研究
通过对HEPT结构改造、研究,A.L.Hopkins对HIV-1逆转录酶晶体的三维结构进行了分析,通过对HEPT及其衍生物与HIV-1逆转录酶作用进行受力、构象及电性作用研究,计算机辅助设计、计算、分析得到如下HEPT构效关系规律〔11〕:1)HEPT类化合物的抗HIV作用主要是它们的6位芳环干扰HIV-1逆转录酶的181位酪氨酸的构象而起作用,干扰越强,则作用越强.2)连接胸腺嘧啶环与苯环结构的硫并不是必需基团,它可以被其它基团如亚甲基等取代.3)HEPT的5位取代基可以干扰HIV-1逆转录酶的181位酪氨酸的构象,如被大体积基团如异丙基取代时,则明显改变181位酪氨酸的构象,而加强6位芳环的干扰作用.4)HEPT与HIV-1逆转录酶的作用区域主要为疏水亲脂环境,对HEPT进行亲脂性改造将会有助于两者的结合.如将HEPT的1位末端羟基连接亲脂基团,将有利于化合物与酶的结合,提高活性.
, 百拇医药
3.2 通过将HIV-1逆转录酶抑制剂与HEPT连接进行构效关系研究
为了进一步研究联合用药的有效性、发展新的抗HIV品种,S.Velazquez将对艾滋病病毒有抑制作用的化合物如:AZT、TSAO等分别用不同长度的碳链与HEPT连接起来,进行药理实验.发现这种连接的产物虽然仍然具有抗HIV-1病毒活性,但其作用已不如连接前两个母体化合物各自的活性,而且对HIV-2无效〔12〕.
3.3 通过对HEPT的电性、亲脂性、空间结构及构象分析进行构效关系研究
M.H.Nguyen等在研究HEPT类化合物构效关系时发现,HEPT的5位乙基或异丙基取代物都要比甲基取代物活性高,如:5-乙基HEPT对HIV-1的EC50为0.1 μmol/L,而5-甲基HEPT的EC50仅为2.6 μmol/L〔3〕,而除去侧链末端亲水的羟基也可以提高化合物的活性〔4〕.J.Renault等认为体积较大的乙基由于空间位阻效应,阻挡了苯环的旋转,而使化合物的活性提高.由此推断将苯环锁住的平面结构可使苯环与胸腺嘧啶环形成大π键共轭体系,可能对提高化合物活性有利.另外经X光衍射晶体结构分析及计算机图形分析发现,HEPT与HIV逆转录酶的作用区主要为亲脂环境,因此设计了一系列五元及六元环结构的化合物将胸腺嘧啶环与苯环锁住,交叉地也把HEPT末端亲水的羟基或用亲脂性的苯环取代,或再增加一个亲水羟基,遗憾地是所合成的化合物中没有一个具有抗HIV活性〔4〕.
, 百拇医药
但是,H.Maruenda等受TIBO和双吡啶类化合物七元环结构的启发,用七元环把胸腺嘧啶环与苯环锁住,并将1位侧链末端亲水的羟基乙醚化以增加化合物的脂溶性,则得到了高活性的衍生物HEPT-25,它的EC50达到0.64 μmol/L〔14〕.
通过合成、药理筛选及构效关系分析,对HEPT类化合物的研究有了新的进展,已有更新、更有效的化合物进入了进一步实验研究,如化合物HEPT-33的EC50达到0.0042 μmol/L,现已进入临床实验阶段〔5〕.
有关HEPT类化合物的合成及生物活性研究工作刚刚起步,方兴未艾.希望在不久的将来,能够有HEPT类的抗病毒新药问世.
, http://www.100md.com
1通讯联系人
参考文献
[1] Balzarini J,Karlsson A,Clercq ED.Human immunodeficiency virus type 1 drug-resistance patterns with different 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine derivative.Molecular Pharmacology,1993,44:694~701
[2] Balzarini J,Baba M,Clercq ED.Differential activities of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine derivatives against different human immunodeficiency virus type 1 mutant strains.Antimicronial Agents and Chemotherapy,1995,39(4):998~1002
, http://www.100md.com
[3] Robert W,Buckheit JR,Valerie FB,et al.Resistance to 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine derivatives is generated by mutations at multiple sites in the HIV-reverse transcriptase.Virology,1995,210:186~193
[4] Renault J,Laduree D,Robba M.Synthesis and antiviral study of dihydrothieno and thianopyrimidine diones acyclic nucleosides as potential anti-HIV agents.Nucleosides & Nucleotides,1994,13(5):1135~1145
[5] Tanaka H,Takashima H,Ubasawa M,et al.Synthesis and antiviral activity of 6-benzyl analogs of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine(HEPT)as potential and selsctive anti-HIV agents.J Med Chem,1995,38:2860~2865
, 百拇医药
[6] Miyasaka T,Tanaka H,Baba M,et al.A novel lead for specific anti-HIV-1 agent:1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine.J Med Chem,1989,32:2507~2509
[7] Tanaka H,Baba M,Hayakawa H,et al.A new class of HIV-1-specific 6-substituted acyclouridine derivatives:synthesis and anti-HIV-1 activity of 5-or 6-substituted analogues of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine(HEPT).J Med Chem,1991,34:349~357
[8] Pontikis R,Monneret C.Synthesis of deoxy analogs of HEPT involving a palladium(o)catalyzed coupling.Tetrahedron Lett,1994,35(25):4351~4354
, http://www.100md.com
[9] Tanaka H,Takashima H,Ubasawa M,et al.Synthesis and antiviral activity of deoxy analogs of 1-〔(2-hydroxyethoxy)methyl〕-6-(phenylthio)thymine(HEPT)as potential and selective anti-HIV agents.J Med Chem,1992,35:4713~4719
[10] Tanaka H,Baba M,Hayakawa H,et al.Lithiation of uracilnucleosides and its application to the synthesis of a new class of anti-HIV-1 acyclonucleosides.Nucleosides & Nucleotides,1991,10(1~3):379~400
[11] Hopkins AL,Ran J,Esnouf RM,et al.Complexes of HIV-1 reverse transcriptase inhibitors of the HEPT series reveal conformational changes relevant to the design of potent non-nucleoside inhibitors.J Med Chem,1996,39:1589~1600
, 百拇医药
[12] Velazqjez S,Alvarez R,San-Felix A,et al.Synthesis and anti-HIV activity of [AZT]-[TSAOT]and [AZT]-[HEPT]dimers as potential multifunctional inhibitors of HIV-1 reverse transcriptase.J Med Chem,1995,38:1641~1649
[13] Nguyen MH,Schinazi RF,Shi C.et al.Resistance of human immunodeficiency virus type 1 to acyclic 6-phenylsenenyl-and phenylthiopyrimidines.Antimicronial Agents and Chemotherapy,1994,38(10):2409~2414
[14] Maruenda H,Johnson F.Design and synthesis of novel inhibitors of HIV-1 reverse transcriptase.J Med Chem,1995,38(12):2145~2151
收稿日期:1998-10-19, 百拇医药(张铭龙 马灵台 张礼和1)