剪切诱导血小板聚集及其机理研究简易厌氧培养皿及厌氧罐检测厌氧菌的对比观察
作者:焦立公 廖福龙
单位:100700 北京市,中国中医研究院中药研究所
关键词:
剪切诱导血小板聚集及其机理研究 导言
在动脉血栓性疾病的病理过程中,血小板活化是重要的始动因素。有许多物质,如凝血酶、肾上腺素等可导致血小板的活化和聚集。近年来研究发现,血流中单纯的高剪切应力也可以诱导血小板聚集。这不但对于存在高剪切应力的血管,如小动脉,动脉痉挛和动脉硬化造成的局部狭窄区的血栓形成有特别重要的意义,还可在其下游形成微血栓,堵塞微血管,造成微循环障碍。因此,研究剪切诱导血小板聚集(Shear induced platelet aggregation,SIPA)的机理有十分重要的意义。
早在1845年由德国病理学家Rudolph Virchow提出血液流动在血栓形成中的作用。他认为,在生理条件下,血液流动、血管因素和血液成分是维持正常凝血、止血功能的三个基本要素。这三个要素中任何一个要素的改变都可导致血栓形成或止血功能障碍。
, http://www.100md.com
血小板在血液循环中沿着血管壁附近的流层流动,不粘附于正常的血管内皮表面。在血管中,靠近管壁的血液流速最低,而在血管轴心部位流速最高。因此,从轴心到管壁形成了不同流层的速度梯度,就是由于这种速度梯度的存在产生了对血小板两个侧面不同的作用力,形成了剪切作用。许多研究的结果支持剪切应力可以直接活化血小板。Moake研究指出:在给予180dynes/cm2的病理性剪切应力0.5和5.0min后,如果有大的von Willebrand因子(vWF)多聚体存在,就出现血小板聚集,而这种聚集不需要其他外源性激活剂。血小板膜糖蛋白Ib/IX(GpIb/IX)和IIb/IIIa(GpIIb/IIIa)的单克隆抗体可以抑制这种聚集。缺乏纤维蛋白原时,则没有多大影响。研究进一步发现,该过程中可观测到血小板内的[Ca2+]升高到原来的10倍以上,EGTA与细胞外Ca2+的鳌合完全抑制剪切应力引起的[Ca2+]升高与血小板聚集;阻断血小板释放ADP也可抑制SIPA[1]。
, http://www.100md.com
这些结果表明剪切诱导的血小板聚集至少有以下因素参与:(1)血小板膜糖蛋白Ib/ΙX 和IIb/IIIa,(2)血浆vWF,(3)一定浓度的血浆Ca2+和ADP。用分子生物学手段研究则对剪切诱导血小板聚集的认识大大深入了。
剪切诱导血小板聚集的分子机制研究
GpIb/IX复合物是血小板膜上最主要的糖蛋白之一。它是由 GpIb和GpIX(17KD)这两个跨膜糖蛋白以1∶1比例通过非共价方式组成的异二聚体复合物,其中GpIb是由两个亚单位GpIbα(145KD)和GpIbβ(24KD)以二硫键连接而成[2]。GpIb/IX的主要功能是识别血管壁中的vWF,使血小板粘附到血管内皮下[3]。对GpIb/IX功能的认识来自于对遗传性巨大血小板综合征(Bernard-soulier syndrome,BSS)患者的临床研究。这种病人临床上呈出血倾向。这种变化可由于GpIbα 、GpIbβ、GpIX或GpV中任何一种的缺失或改变[4]。GpIb/IX与vWF的结合位点,位于GpIbα氨基端的45KD片段中,与GpIbα相当的合成肽丝氨酸251-酪氨酸279和甘氨酸 271-甘氨酸285可抑制血小板与vWF结合[5]。
, http://www.100md.com
GpIIb/IIIa复合物是血小板膜上最丰富的受体,它也存在于血小板表面管道膜和α-颗粒中。当血小板激活时,这些细胞内受体池也能在血小板表面表达。GpIIb/IIa是由GpIIb(136KD)和GpIIIa(92KD)两个亚单位,在钙离子参与下,以1∶1比例通过非共价方式组成的异二聚体复合物[5]。它的主要功能是与纤维蛋白原结合,通过纤维蛋白原的“桥”的作用把血小板与血小板、血小板与内皮下结构等连接起来形成血小板栓。它还可以与其他含有Arg-Gly-Asp(RGD)片段的粘附蛋白如vWF、纤维连接蛋白、外连接蛋白等结合,参与血小板的粘附与聚集。RGD 序列与GpII/IIIa的结合位点在GpIIIa的109-171残基的63个氨基酸的一段序列中。与GpIIb/IIIa结合的另一序列是Lys-Gln-Ala-Asp-Val序列,它只存在于纤维蛋白原中[6]。血小板无力症(Glanzmann’s thrombasthenia)患者血小板缺乏GpIIb/IIIa,临床上呈出血倾向。
, http://www.100md.com vWF在内皮细胞或巨核细胞中合成,在内质网中通过二硫键结合成二聚体,这是vWF的基本构型,还可在高尔基体中形成多聚体。内皮细胞分泌的vWF可进入血液循环,也可进入腔外,从而成为内皮下基质的一部分,在调节血小板粘附到血管壁过程中起重要作用。巨核细胞中的vWF,被包装进入血小板的α-颗粒,当血小板激活时再分泌。在vWF上与GpIb/IX的连接区在Val449-Lys728这么一个52/48KD的片段上。vWF与GpIIb/IIIa的连接区在第1744-1747位氨基酸上,是一个Arg-Gly-Asp-Ser四肽。 vWF通过中介血小板粘附到暴露的血管内皮和促进血小板血栓在血管损伤部位的形成而参与止血过程,特别是有高剪切应力存在的情况下,血小板通过vWF单层覆盖在损伤的内皮下层。在这里vWF作为一个连接“桥”的功能,一端是内皮下成分,如胶原;另一端是血小板GpIb/IX。vWF与GpIb/IX的相互作用引起血小板的活化, 并随着其他激活剂的参与,促进其他血小板补充到生长的血栓上,在这一点上,vWF与GpIIb/IIIa的连接是最重要的。vWF的基因长达150-200KD,任何基因改变影响vWF的合成与表达都可导致vWF质或量的异常,即血管性假血友病(von Willebrand disease,vWD),临床上表现为出血倾向,实验室则表现为血液低凝或不凝[7]。
, 百拇医药
目前的研究资料表明,剪切应力是作用于血小板,而不是作用于血浆中的vWF,使血小板的受体对vWF呈短暂的活化状态。这些对剪切敏感的血小板膜成分大概是GpIb/IX的一部分[8]。人们假设在剪切应力作用下,vWF与GpIb/IX的初始作用成为血小板聚集中血小板“簇”的起始点,并且这个血小板“簇”可以成为一个集中多种血小板活化物质的微环境,包括ADP和肾上腺素等,从而进一步扩大剪切引起的刺激[9]。剪切诱导的初始的触发过程非常难以捉摸,就目前的技术难以研究它微妙的过程。
建立在剪切诱导vWF与GpIb/IX连接基础上的研究资料,支持GpIb/IX与vWF连接后,可发生信号,使血小板外Ca2+转移进入细胞内。另外,抑制vWF与GpIb/IX的连接则可以完全抑制[Ca2+]的升高和聚集反应,而抑制vWF与GpIIb/IIIa 的连接只能部分抑制这两种变化。由此可见,vWF与GpIb/IX的连接对血小板内[Ca2+]的升高和聚集是绝对必要的[1]。vWF与GpIb/IX 连接可开放Ca2+通道的机理尚不明了。但这种细胞内[Ca2+]升高不是简单一个受体被占领的结果,也许还包括以下影响:多聚的vWF分子包括重复的GpIb/IX连接区,它与同一血小板和/或邻近血小板上的几个GpIb/IX 相互连接。研究发现代表vWF上单个GpIb/IX连接的重组片段对剪切诱导的影响是一个有效的抑制剂,但它单独对细胞内[Ca2+]不能产生多大影响,从而支持血小板单个受体占领不能有效地产生Ca2+的跨膜内流[10]。
, 百拇医药
有研究者提出血小板 GpIIb/IIIa 也许就是 Ca2+通道或邻近 Ca2+通道。当用单克隆抗体或合成的RGDS肽抑制配体与GpIIb/IIIa的连接,可引起凝血酶刺激的血小板膜非电压依赖性的Ca2+ 减少。其他资料也显示抑制血小板GpIIb/IIIa上vWF的连接点可抑制(而不是完全消除)细胞内[Ca2+]对剪切应力的反应[1]。也有研究者认为这种作用 更象一种间接的影响,如同在实际中看到的,先天性GpIIb/IIIa缺乏的病人血小板中,剪切诱导的[Ca2+]变化仍正常发生,但不会聚集。支持[Ca2+]变化不是GpIIb/IIIa作用的结果,从而认为这可能依赖于抗体识别的抗原决定簇的定位,在许多情况下,由于空间排列的障碍而使邻近的离子通道的功能受到影响[10]。
通过磷酸肌酸/磷酸肌酸激酶来改变剪切应力下血小板释放ADP的试验,证明ADP对SIPA也是需要的,但它对细胞内[Ca2+]升高没有影响。由此可见剪切应力引起的[Ca2+]升高不是释放的ADP活化血小板的反馈的结果,从而支持剪切应力诱导的 vWF-GpIb相互作用引起的细胞内[Ca2+]升高,早于ADP的释放[1]。由血小板释放的ADP,参与了SIPA,但它不是诱导血小板聚集的主要因素。这一过程中它的具体作用尚不清楚。
, 百拇医药
特异性的第二信使如何把 GpIIb/IIIa变成活化的vWF的受体也不清楚。但是这个信息网可能包括Ca2+、蛋白激酶C(PKC)、丝氨酸-苏氨酸激酶、酪氨酸激酶和磷脂酰肌醇-3-激酶[11]。已知剪切诱导的血小板聚集与血小板―白细胞C激酶底物(pleckstrin)的磷酸化有关,而这种磷酸化完全依赖vWF与血小板受体的连接。血小板-白细胞C激酶底物是PKC的底物,抑制PKC可有效地抑制血小板与vWF相互作用[12,13]。表明当剪切诱导vWF与GpIb/IX连接时,可通过一个使PKC活化的途径使血小板聚集。病理性剪切水平下,血小板酪氨酸激酶也可被活化,但它功能上的重要性仍未确定[14]。
目前已知激活GpIIb/IIIa需细胞外Ca2+的参与,同时至少通过两条途径:一是被G蛋白依赖的肌醇磷脂水解始动,引起PKC激活和[Ca2+]升高;另一条是仅在高浓度凝血酶存在或胶原存在时,不依赖肌醇磷脂的途径[5]。
, http://www.100md.com
剪切诱导血小板聚集过程中钙离子的作用
Ca2+作为第二信使在调节细胞功能中起着极为重要的作用。在剪切诱导血小板聚集过程中Ca2+也起着非常重要的作用。
1 细胞外Ca2+的作用
血小板GPIIb上有4个Ca2+结合位点。GpIIIa可能也有Ca2+结合位点,而且GpIIIa的构型依赖于Ca2+。另外,Ca2+对于维持GpIIb/IIIa复合物的完整性也是必需的。有报道,血小板与2~4mmol/L的EDTA或EGTA在37℃下孵育5~7min,血小板便不可逆地丧失聚集能力。这种变化是由于GpIIb/IIIa复合物不可逆解体所致。
2 细胞外Ca2+的内流和细胞内Ca2+动员
, 百拇医药
血小板受到刺激,可出现细胞外Ca2+内流和细胞内储存的Ca2+的动员,使细胞内Ca2+水平由静息时的60~100nmol/L升至活化后的1000nmol/L[1]。有报道认为这种变化完全是由于细胞外的Ca2+的跨膜进入,这是通过用EGTA或过量的细胞外Mn2+可完全抑制Ca2+的变化和聚集而得出的结论。但也有报道用细胞内Ca2+代谢抑制剂TMB-8也可以抑制血小板的聚集[15]。已知血小板膜上不存在电压依赖性钙通道,因此这种Ca2+内流可能是通过受体调控性钙通道。有关剪切引起的细胞内Ca2+动员了解不多。
3 细胞内Ca2+在血小板活化中的作用
3.1 细胞内Ca2+升高可使肌球蛋白轻链(MLC)发生磷酸化。磷酸化的MLC使肌球蛋白六聚体被肌动蛋白作用,从而引起血小板的形态改变和收缩[16]。
, http://www.100md.com
3.2 有多种Ca2+依赖性蛋白酶在血小板活化中起重要作用。例如,由Ca2+依赖性蛋白酶分解的肌动蛋白结合蛋白和分子量235KD的多肽可引起血小板聚集过程中骨架的重组。
3.3 血小板释放反应必须有Ca2+参加。Ca2+参与血小板内储存颗粒膜和细胞膜的融合,促进胞裂外排发生。
3.4 磷脂酶C(PLC)和磷脂酶A2(PLA2)的活化均受Ca2+的调节。已知PLC可水解血小板磷脂酰肌醇-4.5-二磷酸(PIP2)生成三磷酸肌醇(IP3)和二脂酸甘油(DAG)。IP3可引起细胞内Ca2+增加,而DAG引起PKC活化。这两条途径可各自独立激活血小板,又可协同增强多种血小板的刺激反应。PLA2可作用于不同磷脂底物,生成花生四烯酸(AA)和血小板活化因子(PAF)。
, http://www.100md.com
3.5 蛋白激酶C是引起血小板聚集的重要调节物。已知病理性剪切力可引起不依赖DAG的PKC活化,这种PKC活化需要Ca2+[5]。PKC的活性抑制又可抑制剪切诱导的血小板聚集[13]。PKC还可调节Ca2+依赖性PLA2所介导的AA释放,从而影响血小板活化[16]。
3.6 剪切诱导血小板聚集的最后的、也是最关键的步骤――GPIIb/IIIa与vWF的连接也需要Ca2+的参与[5]。
小结
综上所述,高剪切力下血小板的聚集可通过以下机制形成:首先是高剪切应力下vWF与血小板GpIb/IX的结合,激活了血小板膜上的Ca2+通道,形成了Ca2+跨膜内流。细胞内Ca2+升高作为血小板活化的第二信使,使GpIIb/IIIa活化,成为vWF的受体并与之相连,并通过vWF形成牢固的血小板间联系――聚集[17,18]。如图1[10]。在这里GpIb与vWF的连接相当于激活剂的作用,也许还有启动血小板内在联系的作用。而GpIIb/IIIa与vWF连接则不同,它是形成稳定的血小板聚集所必须的相互作用[10]。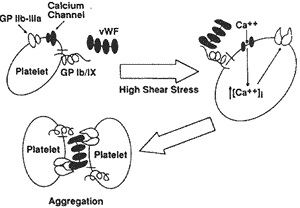
, 百拇医药
图1 剪切诱导血小板聚集机制
(引自参考文献10)
以上所述的有关剪切诱导血小板聚集的试验多是在体外进行、模拟在有完整血管内皮的狭窄动脉中剪切应力直接诱导的血小板聚集,它与内皮损伤起的血小板聚集既有联系又有区别,后者也需要剪切应力的参与,但不需要象前者那样高,一般在20dynes/cm2以下即可。最重要的是它必须有血小板与暴露的血管内皮下成分,如胶原的接触。即以粘附为基础,并在粘附基础上形成血小板聚集,同时还需有纤维蛋白原的参与[18,19]。
但是,在体外进行的剪切诱导的血小板聚集的试验与体内发生的剪切应力对血小板的作用又有一定区别。在体内,在血流动力学外力作用下,可给予内皮细胞以信号,调节其作用于血管和抗血栓物质的合成与释放,而这些物质的主要作用是促进血流[20],如PGI2、内皮松弛因子以及氮的氧化物(NO)及其它血小板抑制剂和纤溶产物[21,22]。通过它们,在内皮完整的情况下抗衡病理性高剪切应力的促凝作用,但病理性高剪切应力产生的促凝作用和通过完整的内皮细胞产生的抗凝作用如何保持平衡,以及这种平衡在什么条件下又如何被打破则有待进一步研究。
, http://www.100md.com
作者简介:焦立公 综述
廖福龙 审校
参考文献
1 Chew TW.Shear stress-induced vWF binding to platelet GpIb initiates with aggregation.Blood.1992,80:113
2 Peerschke EIB.Platelet membranes and receptors.Thrombosis and Hemorrhage.1994,1:219
3 Harvcy R Gralnick.Platelet adhesion and high shear rates:the roles of vWF/GPIb and the β1 integrin α2β1.Thrombosis Research.1996,81:13
, http://www.100md.com
4 Handin RI.Platelet membrane proteins and their disorders.Blood.1995,90:1049
5 江钟,郑植荃.现代血栓病学.北京:北京医科大学中国协和医科大学联合出版社.1997,10
6 武艺.心血管疾病血小板糖蛋白IIb/IIIa受体的研究进展.国外医学心血管疾病分册.1996,23:151
7 Edith Fressinaud.Shear rate-dependent impairment of thrombus growth on collagen in nonanticogulated blood from patients with von Willebrand disease and hemophilia.Blood.1992,80:988
, 百拇医药 8 Lopez JA.The platelet glycoprotein Ib-IX complex.Blood Coagul Fibrin.1994,5:97
9 Goto S.Characterization of the unique mechanisms mediating the shear-dependent binding of soluble von Willebrand factor to platelets.J Biol Chem.1995,270:233
10 Ikcda K.Transmcmbranc calcium influx associated with vWF binding to GpIb in the initiation of shear -induced platelet aggregation.Thromb Haeomost.1993,69:496
, 百拇医药 11 Kovacsovics TJ.Phosphoinositide 3-Kinasc inhibition spares actin assembly in activating platelet but reverses platelet aggregation.J Biol Chem.1995,270:113
12 Ruggcri ZM.New insights into the mechanisms of platelet adhesion and aggregation.Scmin Hematol.1994,31:229
13 Kroll MH.Protein Kinasc C is activated in platelets subjected to patholgical shear stress.J Biol Chem.1993,268:352
14 Razdan K.Shear stress-induced von Willebrand factor binding to platelets causes the activation of tyrosine kinase(s).Biochem J.1994,302:681
, http://www.100md.com
15 P Dandona.Calicium,Calmoduline and protein Kinasc C dependence of platelet shape change.Thrombosis Research.1996,81:132
16 王美健.钙离子与血小板活化.中华血液学杂志.1992,13:268
17 Goto S.Characterization of the unique mechanism mediating the shear dependent binding of solublc vWF to platelet.J Biol Chem.1995,270:233
18 Ruggeri ZM.Mechanisms of shear-induced platelet adhesion and aggregation.Thromb Haemost.1993,70:119
, 百拇医药
19 Ruggeri ZM.GpIb and vWF in the process of thrombus formation.Ann N Y Acad Sci.1994,714:200
20 Madeleine M Mazeaud.Platelet aggregation and in vivo shear forces.Thrombosis and Haemostasis.1994,71:26
21 Kolle A.Role of shear stress and endothelial prostaglandins in flowand viscosityinduced dilation of arterioles in vitro.Circ Rcs.1993,72:1276
22 Randall ME.EDRF plays central role in collateral flow after arterial occlasion in rabbit ear.Am J Physiol.1992,263:H752
收稿:1998-04-08 修回:1998-07-03
中国微生态学杂志 1999年第4期第11卷 技术与方法
简易厌氧培养皿及厌氧罐检测厌氧菌的对比观察
作者:朱 江 孔繁林 储从家 罗次节
单位:云南玉溪市人民医院检验科 653100
关键词:, 百拇医药
单位:100700 北京市,中国中医研究院中药研究所
关键词:
剪切诱导血小板聚集及其机理研究 导言
在动脉血栓性疾病的病理过程中,血小板活化是重要的始动因素。有许多物质,如凝血酶、肾上腺素等可导致血小板的活化和聚集。近年来研究发现,血流中单纯的高剪切应力也可以诱导血小板聚集。这不但对于存在高剪切应力的血管,如小动脉,动脉痉挛和动脉硬化造成的局部狭窄区的血栓形成有特别重要的意义,还可在其下游形成微血栓,堵塞微血管,造成微循环障碍。因此,研究剪切诱导血小板聚集(Shear induced platelet aggregation,SIPA)的机理有十分重要的意义。
早在1845年由德国病理学家Rudolph Virchow提出血液流动在血栓形成中的作用。他认为,在生理条件下,血液流动、血管因素和血液成分是维持正常凝血、止血功能的三个基本要素。这三个要素中任何一个要素的改变都可导致血栓形成或止血功能障碍。
, http://www.100md.com
血小板在血液循环中沿着血管壁附近的流层流动,不粘附于正常的血管内皮表面。在血管中,靠近管壁的血液流速最低,而在血管轴心部位流速最高。因此,从轴心到管壁形成了不同流层的速度梯度,就是由于这种速度梯度的存在产生了对血小板两个侧面不同的作用力,形成了剪切作用。许多研究的结果支持剪切应力可以直接活化血小板。Moake研究指出:在给予180dynes/cm2的病理性剪切应力0.5和5.0min后,如果有大的von Willebrand因子(vWF)多聚体存在,就出现血小板聚集,而这种聚集不需要其他外源性激活剂。血小板膜糖蛋白Ib/IX(GpIb/IX)和IIb/IIIa(GpIIb/IIIa)的单克隆抗体可以抑制这种聚集。缺乏纤维蛋白原时,则没有多大影响。研究进一步发现,该过程中可观测到血小板内的[Ca2+]升高到原来的10倍以上,EGTA与细胞外Ca2+的鳌合完全抑制剪切应力引起的[Ca2+]升高与血小板聚集;阻断血小板释放ADP也可抑制SIPA[1]。
, http://www.100md.com
这些结果表明剪切诱导的血小板聚集至少有以下因素参与:(1)血小板膜糖蛋白Ib/ΙX 和IIb/IIIa,(2)血浆vWF,(3)一定浓度的血浆Ca2+和ADP。用分子生物学手段研究则对剪切诱导血小板聚集的认识大大深入了。
剪切诱导血小板聚集的分子机制研究
GpIb/IX复合物是血小板膜上最主要的糖蛋白之一。它是由 GpIb和GpIX(17KD)这两个跨膜糖蛋白以1∶1比例通过非共价方式组成的异二聚体复合物,其中GpIb是由两个亚单位GpIbα(145KD)和GpIbβ(24KD)以二硫键连接而成[2]。GpIb/IX的主要功能是识别血管壁中的vWF,使血小板粘附到血管内皮下[3]。对GpIb/IX功能的认识来自于对遗传性巨大血小板综合征(Bernard-soulier syndrome,BSS)患者的临床研究。这种病人临床上呈出血倾向。这种变化可由于GpIbα 、GpIbβ、GpIX或GpV中任何一种的缺失或改变[4]。GpIb/IX与vWF的结合位点,位于GpIbα氨基端的45KD片段中,与GpIbα相当的合成肽丝氨酸251-酪氨酸279和甘氨酸 271-甘氨酸285可抑制血小板与vWF结合[5]。
, http://www.100md.com
GpIIb/IIIa复合物是血小板膜上最丰富的受体,它也存在于血小板表面管道膜和α-颗粒中。当血小板激活时,这些细胞内受体池也能在血小板表面表达。GpIIb/IIa是由GpIIb(136KD)和GpIIIa(92KD)两个亚单位,在钙离子参与下,以1∶1比例通过非共价方式组成的异二聚体复合物[5]。它的主要功能是与纤维蛋白原结合,通过纤维蛋白原的“桥”的作用把血小板与血小板、血小板与内皮下结构等连接起来形成血小板栓。它还可以与其他含有Arg-Gly-Asp(RGD)片段的粘附蛋白如vWF、纤维连接蛋白、外连接蛋白等结合,参与血小板的粘附与聚集。RGD 序列与GpII/IIIa的结合位点在GpIIIa的109-171残基的63个氨基酸的一段序列中。与GpIIb/IIIa结合的另一序列是Lys-Gln-Ala-Asp-Val序列,它只存在于纤维蛋白原中[6]。血小板无力症(Glanzmann’s thrombasthenia)患者血小板缺乏GpIIb/IIIa,临床上呈出血倾向。
, http://www.100md.com vWF在内皮细胞或巨核细胞中合成,在内质网中通过二硫键结合成二聚体,这是vWF的基本构型,还可在高尔基体中形成多聚体。内皮细胞分泌的vWF可进入血液循环,也可进入腔外,从而成为内皮下基质的一部分,在调节血小板粘附到血管壁过程中起重要作用。巨核细胞中的vWF,被包装进入血小板的α-颗粒,当血小板激活时再分泌。在vWF上与GpIb/IX的连接区在Val449-Lys728这么一个52/48KD的片段上。vWF与GpIIb/IIIa的连接区在第1744-1747位氨基酸上,是一个Arg-Gly-Asp-Ser四肽。 vWF通过中介血小板粘附到暴露的血管内皮和促进血小板血栓在血管损伤部位的形成而参与止血过程,特别是有高剪切应力存在的情况下,血小板通过vWF单层覆盖在损伤的内皮下层。在这里vWF作为一个连接“桥”的功能,一端是内皮下成分,如胶原;另一端是血小板GpIb/IX。vWF与GpIb/IX的相互作用引起血小板的活化, 并随着其他激活剂的参与,促进其他血小板补充到生长的血栓上,在这一点上,vWF与GpIIb/IIIa的连接是最重要的。vWF的基因长达150-200KD,任何基因改变影响vWF的合成与表达都可导致vWF质或量的异常,即血管性假血友病(von Willebrand disease,vWD),临床上表现为出血倾向,实验室则表现为血液低凝或不凝[7]。
, 百拇医药
目前的研究资料表明,剪切应力是作用于血小板,而不是作用于血浆中的vWF,使血小板的受体对vWF呈短暂的活化状态。这些对剪切敏感的血小板膜成分大概是GpIb/IX的一部分[8]。人们假设在剪切应力作用下,vWF与GpIb/IX的初始作用成为血小板聚集中血小板“簇”的起始点,并且这个血小板“簇”可以成为一个集中多种血小板活化物质的微环境,包括ADP和肾上腺素等,从而进一步扩大剪切引起的刺激[9]。剪切诱导的初始的触发过程非常难以捉摸,就目前的技术难以研究它微妙的过程。
建立在剪切诱导vWF与GpIb/IX连接基础上的研究资料,支持GpIb/IX与vWF连接后,可发生信号,使血小板外Ca2+转移进入细胞内。另外,抑制vWF与GpIb/IX的连接则可以完全抑制[Ca2+]的升高和聚集反应,而抑制vWF与GpIIb/IIIa 的连接只能部分抑制这两种变化。由此可见,vWF与GpIb/IX的连接对血小板内[Ca2+]的升高和聚集是绝对必要的[1]。vWF与GpIb/IX 连接可开放Ca2+通道的机理尚不明了。但这种细胞内[Ca2+]升高不是简单一个受体被占领的结果,也许还包括以下影响:多聚的vWF分子包括重复的GpIb/IX连接区,它与同一血小板和/或邻近血小板上的几个GpIb/IX 相互连接。研究发现代表vWF上单个GpIb/IX连接的重组片段对剪切诱导的影响是一个有效的抑制剂,但它单独对细胞内[Ca2+]不能产生多大影响,从而支持血小板单个受体占领不能有效地产生Ca2+的跨膜内流[10]。
, 百拇医药
有研究者提出血小板 GpIIb/IIIa 也许就是 Ca2+通道或邻近 Ca2+通道。当用单克隆抗体或合成的RGDS肽抑制配体与GpIIb/IIIa的连接,可引起凝血酶刺激的血小板膜非电压依赖性的Ca2+ 减少。其他资料也显示抑制血小板GpIIb/IIIa上vWF的连接点可抑制(而不是完全消除)细胞内[Ca2+]对剪切应力的反应[1]。也有研究者认为这种作用 更象一种间接的影响,如同在实际中看到的,先天性GpIIb/IIIa缺乏的病人血小板中,剪切诱导的[Ca2+]变化仍正常发生,但不会聚集。支持[Ca2+]变化不是GpIIb/IIIa作用的结果,从而认为这可能依赖于抗体识别的抗原决定簇的定位,在许多情况下,由于空间排列的障碍而使邻近的离子通道的功能受到影响[10]。
通过磷酸肌酸/磷酸肌酸激酶来改变剪切应力下血小板释放ADP的试验,证明ADP对SIPA也是需要的,但它对细胞内[Ca2+]升高没有影响。由此可见剪切应力引起的[Ca2+]升高不是释放的ADP活化血小板的反馈的结果,从而支持剪切应力诱导的 vWF-GpIb相互作用引起的细胞内[Ca2+]升高,早于ADP的释放[1]。由血小板释放的ADP,参与了SIPA,但它不是诱导血小板聚集的主要因素。这一过程中它的具体作用尚不清楚。
, 百拇医药
特异性的第二信使如何把 GpIIb/IIIa变成活化的vWF的受体也不清楚。但是这个信息网可能包括Ca2+、蛋白激酶C(PKC)、丝氨酸-苏氨酸激酶、酪氨酸激酶和磷脂酰肌醇-3-激酶[11]。已知剪切诱导的血小板聚集与血小板―白细胞C激酶底物(pleckstrin)的磷酸化有关,而这种磷酸化完全依赖vWF与血小板受体的连接。血小板-白细胞C激酶底物是PKC的底物,抑制PKC可有效地抑制血小板与vWF相互作用[12,13]。表明当剪切诱导vWF与GpIb/IX连接时,可通过一个使PKC活化的途径使血小板聚集。病理性剪切水平下,血小板酪氨酸激酶也可被活化,但它功能上的重要性仍未确定[14]。
目前已知激活GpIIb/IIIa需细胞外Ca2+的参与,同时至少通过两条途径:一是被G蛋白依赖的肌醇磷脂水解始动,引起PKC激活和[Ca2+]升高;另一条是仅在高浓度凝血酶存在或胶原存在时,不依赖肌醇磷脂的途径[5]。
, http://www.100md.com
剪切诱导血小板聚集过程中钙离子的作用
Ca2+作为第二信使在调节细胞功能中起着极为重要的作用。在剪切诱导血小板聚集过程中Ca2+也起着非常重要的作用。
1 细胞外Ca2+的作用
血小板GPIIb上有4个Ca2+结合位点。GpIIIa可能也有Ca2+结合位点,而且GpIIIa的构型依赖于Ca2+。另外,Ca2+对于维持GpIIb/IIIa复合物的完整性也是必需的。有报道,血小板与2~4mmol/L的EDTA或EGTA在37℃下孵育5~7min,血小板便不可逆地丧失聚集能力。这种变化是由于GpIIb/IIIa复合物不可逆解体所致。
2 细胞外Ca2+的内流和细胞内Ca2+动员
, 百拇医药
血小板受到刺激,可出现细胞外Ca2+内流和细胞内储存的Ca2+的动员,使细胞内Ca2+水平由静息时的60~100nmol/L升至活化后的1000nmol/L[1]。有报道认为这种变化完全是由于细胞外的Ca2+的跨膜进入,这是通过用EGTA或过量的细胞外Mn2+可完全抑制Ca2+的变化和聚集而得出的结论。但也有报道用细胞内Ca2+代谢抑制剂TMB-8也可以抑制血小板的聚集[15]。已知血小板膜上不存在电压依赖性钙通道,因此这种Ca2+内流可能是通过受体调控性钙通道。有关剪切引起的细胞内Ca2+动员了解不多。
3 细胞内Ca2+在血小板活化中的作用
3.1 细胞内Ca2+升高可使肌球蛋白轻链(MLC)发生磷酸化。磷酸化的MLC使肌球蛋白六聚体被肌动蛋白作用,从而引起血小板的形态改变和收缩[16]。
, http://www.100md.com
3.2 有多种Ca2+依赖性蛋白酶在血小板活化中起重要作用。例如,由Ca2+依赖性蛋白酶分解的肌动蛋白结合蛋白和分子量235KD的多肽可引起血小板聚集过程中骨架的重组。
3.3 血小板释放反应必须有Ca2+参加。Ca2+参与血小板内储存颗粒膜和细胞膜的融合,促进胞裂外排发生。
3.4 磷脂酶C(PLC)和磷脂酶A2(PLA2)的活化均受Ca2+的调节。已知PLC可水解血小板磷脂酰肌醇-4.5-二磷酸(PIP2)生成三磷酸肌醇(IP3)和二脂酸甘油(DAG)。IP3可引起细胞内Ca2+增加,而DAG引起PKC活化。这两条途径可各自独立激活血小板,又可协同增强多种血小板的刺激反应。PLA2可作用于不同磷脂底物,生成花生四烯酸(AA)和血小板活化因子(PAF)。
, http://www.100md.com
3.5 蛋白激酶C是引起血小板聚集的重要调节物。已知病理性剪切力可引起不依赖DAG的PKC活化,这种PKC活化需要Ca2+[5]。PKC的活性抑制又可抑制剪切诱导的血小板聚集[13]。PKC还可调节Ca2+依赖性PLA2所介导的AA释放,从而影响血小板活化[16]。
3.6 剪切诱导血小板聚集的最后的、也是最关键的步骤――GPIIb/IIIa与vWF的连接也需要Ca2+的参与[5]。
小结
综上所述,高剪切力下血小板的聚集可通过以下机制形成:首先是高剪切应力下vWF与血小板GpIb/IX的结合,激活了血小板膜上的Ca2+通道,形成了Ca2+跨膜内流。细胞内Ca2+升高作为血小板活化的第二信使,使GpIIb/IIIa活化,成为vWF的受体并与之相连,并通过vWF形成牢固的血小板间联系――聚集[17,18]。如图1[10]。在这里GpIb与vWF的连接相当于激活剂的作用,也许还有启动血小板内在联系的作用。而GpIIb/IIIa与vWF连接则不同,它是形成稳定的血小板聚集所必须的相互作用[10]。
, 百拇医药
图1 剪切诱导血小板聚集机制
(引自参考文献10)
以上所述的有关剪切诱导血小板聚集的试验多是在体外进行、模拟在有完整血管内皮的狭窄动脉中剪切应力直接诱导的血小板聚集,它与内皮损伤起的血小板聚集既有联系又有区别,后者也需要剪切应力的参与,但不需要象前者那样高,一般在20dynes/cm2以下即可。最重要的是它必须有血小板与暴露的血管内皮下成分,如胶原的接触。即以粘附为基础,并在粘附基础上形成血小板聚集,同时还需有纤维蛋白原的参与[18,19]。
但是,在体外进行的剪切诱导的血小板聚集的试验与体内发生的剪切应力对血小板的作用又有一定区别。在体内,在血流动力学外力作用下,可给予内皮细胞以信号,调节其作用于血管和抗血栓物质的合成与释放,而这些物质的主要作用是促进血流[20],如PGI2、内皮松弛因子以及氮的氧化物(NO)及其它血小板抑制剂和纤溶产物[21,22]。通过它们,在内皮完整的情况下抗衡病理性高剪切应力的促凝作用,但病理性高剪切应力产生的促凝作用和通过完整的内皮细胞产生的抗凝作用如何保持平衡,以及这种平衡在什么条件下又如何被打破则有待进一步研究。
, http://www.100md.com
作者简介:焦立公 综述
廖福龙 审校
参考文献
1 Chew TW.Shear stress-induced vWF binding to platelet GpIb initiates with aggregation.Blood.1992,80:113
2 Peerschke EIB.Platelet membranes and receptors.Thrombosis and Hemorrhage.1994,1:219
3 Harvcy R Gralnick.Platelet adhesion and high shear rates:the roles of vWF/GPIb and the β1 integrin α2β1.Thrombosis Research.1996,81:13
, http://www.100md.com
4 Handin RI.Platelet membrane proteins and their disorders.Blood.1995,90:1049
5 江钟,郑植荃.现代血栓病学.北京:北京医科大学中国协和医科大学联合出版社.1997,10
6 武艺.心血管疾病血小板糖蛋白IIb/IIIa受体的研究进展.国外医学心血管疾病分册.1996,23:151
7 Edith Fressinaud.Shear rate-dependent impairment of thrombus growth on collagen in nonanticogulated blood from patients with von Willebrand disease and hemophilia.Blood.1992,80:988
, 百拇医药 8 Lopez JA.The platelet glycoprotein Ib-IX complex.Blood Coagul Fibrin.1994,5:97
9 Goto S.Characterization of the unique mechanisms mediating the shear-dependent binding of soluble von Willebrand factor to platelets.J Biol Chem.1995,270:233
10 Ikcda K.Transmcmbranc calcium influx associated with vWF binding to GpIb in the initiation of shear -induced platelet aggregation.Thromb Haeomost.1993,69:496
, 百拇医药 11 Kovacsovics TJ.Phosphoinositide 3-Kinasc inhibition spares actin assembly in activating platelet but reverses platelet aggregation.J Biol Chem.1995,270:113
12 Ruggcri ZM.New insights into the mechanisms of platelet adhesion and aggregation.Scmin Hematol.1994,31:229
13 Kroll MH.Protein Kinasc C is activated in platelets subjected to patholgical shear stress.J Biol Chem.1993,268:352
14 Razdan K.Shear stress-induced von Willebrand factor binding to platelets causes the activation of tyrosine kinase(s).Biochem J.1994,302:681
, http://www.100md.com
15 P Dandona.Calicium,Calmoduline and protein Kinasc C dependence of platelet shape change.Thrombosis Research.1996,81:132
16 王美健.钙离子与血小板活化.中华血液学杂志.1992,13:268
17 Goto S.Characterization of the unique mechanism mediating the shear dependent binding of solublc vWF to platelet.J Biol Chem.1995,270:233
18 Ruggeri ZM.Mechanisms of shear-induced platelet adhesion and aggregation.Thromb Haemost.1993,70:119
, 百拇医药
19 Ruggeri ZM.GpIb and vWF in the process of thrombus formation.Ann N Y Acad Sci.1994,714:200
20 Madeleine M Mazeaud.Platelet aggregation and in vivo shear forces.Thrombosis and Haemostasis.1994,71:26
21 Kolle A.Role of shear stress and endothelial prostaglandins in flowand viscosityinduced dilation of arterioles in vitro.Circ Rcs.1993,72:1276
22 Randall ME.EDRF plays central role in collateral flow after arterial occlasion in rabbit ear.Am J Physiol.1992,263:H752
收稿:1998-04-08 修回:1998-07-03
中国微生态学杂志 1999年第4期第11卷 技术与方法
简易厌氧培养皿及厌氧罐检测厌氧菌的对比观察
作者:朱 江 孔繁林 储从家 罗次节
单位:云南玉溪市人民医院检验科 653100
关键词:, 百拇医药