环氧合酶-2抑制剂的对接(Dock)研究1
作者:朱七庆 屈凌波 雷新胜 郭宗儒2
单位:中国医学科学院 中国协和医科大学药物研究所,北京 100050
关键词:环氧合酶-2;构效关系;分子图形学;对接
摘 要 环氧合酶摘 要 环氧合酶-2具有多种结构类型的抑制剂,比较分子力场分析难以建立它们的定量构效关系.根据自由能原理,抑制剂结合酶的自由能变与活性呈线性关系,本文选择了12个典型结构的的环氧合酶-2抑制剂进行对接,通过分析结合能与活性的相关性,得到了较好的构效模型,相关系数r=0.857,在建立构效模型的同时,通过图形学分析,揭示了这些抑制剂具有较高活性的原因.
Study on the Dock of Cyclooxygenase-2 Inhibitors
Zhu Qiqing,Qu Lingbo,Lei Xinsheng,Guo Zongru
, 百拇医药
Institute of Materia Medica,Chinese Academy of Medical Sciences
and Peking Union Medical College,Beijing 100050
Abstract Selective cyclooxygenase-2(COX-2) inhibitors contain varions structure skeletons.It is more difficult to construct their three dimensional quantitative structure-activity relationship by using the comparative molecular field analysis.In order to bulid their QSAR model and guide the design of a new inhibitor,in this thesis,12 selective COX-2 inhibitor were selected to dock into COX-2 enzyme by using the Dock program.The results showed that there existed a good correlation between activity-logIC50 and binding energy,the relative coefficient r=0.857.In addition Dock mimiced the binding of NS-398 analogueed to COX-2,and the results indicated that NS-398 analogues possessed higher activity because of the presence of additional hydrogen-bonding.
, 百拇医药
Key words cyclooxygenase-2;QSAR molecular graphics;Dock
研究环氧合酶-2(COX-2)特异性抑制剂,是创新新型抗炎药物的重要内容.对于三环类抑制剂,通过比较分子力场分析,已经建立了有效的三维定量构效关系模型.然而,在已有的环氧合酶抑制剂中,除了三环类化合物外,双环类化合物尼美舒利(Nimisulide)和NS-398〔1〕等也是典型的环氧合酶-2抑制剂,这些化合物结构差别较大,分子中起主要或次要作用的力场在三维空间中有可能发生位置上的改变.对于整个分子而言,由于空间性质上的涨落现象,这种场的变化未必会造成分子活性的降低,但是采用原有的叠合方式进行力场比较已经失去意义,使用CoMFA,回归力场与活性的相关性分析往往难以得到有效的QSAR方程.
由于环氧合酶-2的晶体结构已经确定,并已知与抑制剂的结合位点〔2,3〕,本文利用对接程序(Dock),通过计算系列抑制剂与环氧合酶-2的结合能,建立结构变化较大的抑制剂与活性之间的相关方程,同时探求不同结构类型抑制剂结合环氧合酶-2的特征,为环氧合酶-2抑制剂的设计提供有关结构信息.
, 百拇医药
所有计算工作都是在中国医学科学院 中国协和医科大学药物研究所合成室工作站Silicon Graphics IRIS Indigo R40000计算机工作站上完成,应用程序为Tripos公司最新提供的SYBYL 6.4分子软件包〔4〕。计算过程中除特别指明外,所选用的参数均为缺省值,环氧合酶-2与抑制剂的复合物晶体结构从PDB数据库中获得〔5〕.
1 计算方法
1.1 基本原理
药物分子发挥药效必须结合受体形成复合物,生物效应经构成复合物的药物的内在活性引发,药物的作用强度与形成复合物的数目相关,复合物的数目越多,药物的药理作用就越强〔6〕.该过程可简单描述为:]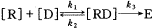 ,[R]为游离受体分子浓度,[D]为游离药物分子浓度,[RD]为药物-受体复合物浓度,E为生物效应.由此可知,药物与受体结合是一个化学平衡,复合物的数目与该平衡的平衡常数K=k1/k2有关.根据热力学原理,在等温等压条件下,系统自由能变ΔG与平衡常数K的关系为:ΔG=-2.303RTlogK 〈1〉,而根据自由能方程:ΔG=ΔH-TΔS 〈2〉,一系列抑制剂与同一受体结合过程中,假定熵变为一常数,自由能变ΔG与焓变ΔH相等,而焓变近似等于系统内能的变化,配体与受体的结合能Eb定义为系统自由能变ΔG,于是可得方程:ΔEb=ΔG=ΔH=ΔE=Ecomplex-ER-EL 〈3〉,Ecomplex为复合物的能量,ER为游离受体的能量,EL为配体的药效构象能.又由于药效与平稳常数的关系为:药效=B*K*IC50 〈4〉,B是比例因子,K为平衡常数,在测定某个系列化合物的活性时,式〈4〉的左侧是常数,因此-logIC50与logK线性相关,而logK又与ΔG线性相关,于是-logIC50与结合能Eb线性相关.显然,通过回归分析-logIC50与Eb相关性可建立如下具有预测能力的关系式:-logIC50=aEb+c 〈5〉,对接程序(Dock)利用经典分子力学原理通过优化受体和配体的相互作用,计算出复合物的能量,并模拟药物分子与受体分子的结合.因此,通过对接操作,根据自由能原理,将可建立-logIC50与结合能Eb的相关方程.
,[R]为游离受体分子浓度,[D]为游离药物分子浓度,[RD]为药物-受体复合物浓度,E为生物效应.由此可知,药物与受体结合是一个化学平衡,复合物的数目与该平衡的平衡常数K=k1/k2有关.根据热力学原理,在等温等压条件下,系统自由能变ΔG与平衡常数K的关系为:ΔG=-2.303RTlogK 〈1〉,而根据自由能方程:ΔG=ΔH-TΔS 〈2〉,一系列抑制剂与同一受体结合过程中,假定熵变为一常数,自由能变ΔG与焓变ΔH相等,而焓变近似等于系统内能的变化,配体与受体的结合能Eb定义为系统自由能变ΔG,于是可得方程:ΔEb=ΔG=ΔH=ΔE=Ecomplex-ER-EL 〈3〉,Ecomplex为复合物的能量,ER为游离受体的能量,EL为配体的药效构象能.又由于药效与平稳常数的关系为:药效=B*K*IC50 〈4〉,B是比例因子,K为平衡常数,在测定某个系列化合物的活性时,式〈4〉的左侧是常数,因此-logIC50与logK线性相关,而logK又与ΔG线性相关,于是-logIC50与结合能Eb线性相关.显然,通过回归分析-logIC50与Eb相关性可建立如下具有预测能力的关系式:-logIC50=aEb+c 〈5〉,对接程序(Dock)利用经典分子力学原理通过优化受体和配体的相互作用,计算出复合物的能量,并模拟药物分子与受体分子的结合.因此,通过对接操作,根据自由能原理,将可建立-logIC50与结合能Eb的相关方程.
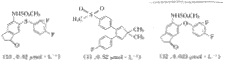
, 百拇医药
Fig.1 The compounds used for Dock analysis
1.2 对接化合物的选择
进行对接研究的12个活性已知但结构不同的化合物结构如图1所示.这12个化合物根据亚结构特征分属于三种不同的结构类型,即三环类、双环类和吲哚美辛.考虑到对接分子的多样性,虽然吲哚美辛对于环氧合酶-2的选择性不高,但其结合COX-2的能力较高并且结构变化更大,因此选其为对接分子.所有12个化合物的活性都具有相同的测试标准,活性之间具有可参比性,活性IC50值列于图1中.
三环类的NC-398和吲哚美辛结合受体的晶体结构的解析为该类化合物的活性构象确立了实验基础(2,7~9).NC-398类化合物虽然与三环系化合物的结构有所区别,但是,药效团的分布是相似的,本文通过代表性的抑制剂NS-398的有限制条件的构象搜索,来寻找该类抑制剂的活性构象.
, http://www.100md.com
1.3 NS-398类化合物的构象分析
NS-398类化合物与三环系化合物的最大区别在于缺少联结两个芳环的中间环,醚键相连的两个苯环与砜基则与三环系中的两个芳环和砜基相对应.显然,NS-398中的这些基团在三维空间的配置应与三环系化合物一致,这两类化合物理应有相同的结合位点.如图2所示,以SC-558为例,定义该类抑制剂的药效团为砜基的硫原子(C)以及两个苯环中心(A)和(B),三个药效团原子间的距离分别表示为d1,d\-2,d\-3,在SC-558活性构象中,d1,d2,d3分别为6.993 ,4.696,3.177,这将作为NS-398构象搜索的限定条件.由于在构象搜索时单键的旋转并不影响S-298中A,B之间和B,C之间的距离变化,因此仅以d1作为限制条件对NS-398类化合物的构象进行系统搜索.NS-398的起始构象以Sybyl 6.4中的“Build/Edit”构建,d1距离限制的分辨率为d1=6.993±0.2,四个旋转单键如图2所示,旋转步长为30°.在进行系统搜索时,同时计算构象能但不进行能量优化,计算使用Tripos力场,原子电荷为PM3计算的MOPAC电荷,最大能差设为10.0 kcal/mol,因此在搜索过程中能量大于最低能量10 kcal/mol的构象将被删除.对所得构象最后都进行构型优化,最小化选用Minmize 2分子力学程序,力场为Tripos力场,能量收敛限为0.05 kcal/mol.最后以SC-558分子为模板,通过分子叠合二者的最大相似性,为NS-类化合物对接确定药效构象.
,4.696,3.177,这将作为NS-398构象搜索的限定条件.由于在构象搜索时单键的旋转并不影响S-298中A,B之间和B,C之间的距离变化,因此仅以d1作为限制条件对NS-398类化合物的构象进行系统搜索.NS-398的起始构象以Sybyl 6.4中的“Build/Edit”构建,d1距离限制的分辨率为d1=6.993±0.2,四个旋转单键如图2所示,旋转步长为30°.在进行系统搜索时,同时计算构象能但不进行能量优化,计算使用Tripos力场,原子电荷为PM3计算的MOPAC电荷,最大能差设为10.0 kcal/mol,因此在搜索过程中能量大于最低能量10 kcal/mol的构象将被删除.对所得构象最后都进行构型优化,最小化选用Minmize 2分子力学程序,力场为Tripos力场,能量收敛限为0.05 kcal/mol.最后以SC-558分子为模板,通过分子叠合二者的最大相似性,为NS-类化合物对接确定药效构象.
, 百拇医药
Fig.2 Distance constraints used for conformational search
1.4 已知化合物的对接及构效方程的建立
对接操作的关键步骤是确定配体分子的初始位置.三类已知抑制剂中,三环类抑制剂的定位以复合物中配体分子SC-558为模板进行分子叠合定位,吲哚美辛的晶体结构则是已知的,经过加氢、荷载电荷直接可以进行对接.NS-398类化合物的定位则通过它的活性构象与三环系的分子叠合来进行.对接过程中选用Tripos力场优化配体和受体之间的相互作用,COX-2荷载Kollman all-atom电荷,配体分子带PM3计算的MOPAC电荷,介电常数与距离相关ε=4rij,能量收敛限为0.1 kcal/mol,计算仅考虑配体分子4范围内的氨基酸残基与配体的相互作用.整个对接过程分两步进行,第一步,固定受体COX-2的几何构型,仅优化配体分子;第二步,对配体分子12范围内的氨基酸不做任何限制,再次优化配体和受体之间的相互作用直至能量收敛.最后记录每次对接后配体与受体的相互作用能Eint和复合物的能量值Ecomplex,利用非交叉验证PLS方法回归分析活性与结合能的相关性,建立定量构效方程.
, 百拇医药
2 结果与讨论
以d1作为限制条件,经系统搜索得到10个NS-398的低能构象,这10个构象两两互成镜像,镜像分子具有相同的能量值.结构优化后,属于镜像同侧的5个构象体都属于相同的几何构型,因此,最终仅得2个分子构象,分别记为构象a和构象b,构象a中环己烷环朝内,构象b的环己烷环朝外.这两个构象与能量最低构象几乎具有相同的几何形状,主要区别在于磺酰胺侧链的方向不同,其能量差小于3.0 kcal/mol.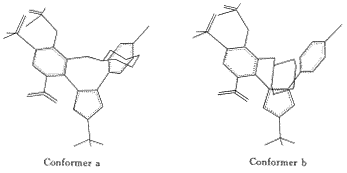
Fig.3 Superimposition of Conformer a,b on SC-558
根据构象分析结果,认为NS-398类化合物应以构象a或构象b与COX-2结合.由于三环系分子的活性构象是已知的,NS-398类与三环系化合物具有相同的药效团,因此通过构象a和构象b与三环系化合物SC-558活性构象的叠合,可确定NS-398的活性构象,两种构象叠合后如图4所示,构象a的环己烷环与SC-558的另一个苯环重叠,构象b的环己烷却远离苯环,由叠合图可知,构象a与SC-558活性构象具有更好的相似性,构象a可以作为NS-398类抑制剂的活性构象,以该叠合后的位置作为对接NS-398类抑制剂对接时的初始位置,构象a中距离d-1,d-2,d-3分别为6.92,4.84,3.78.
, 百拇医药
表1列出了12个化合物对接后的能量项,Eint为对接后配体和受体的相互作用能,Este和Eele分别为配体和受体相互作用能中的立体相互作用能和静电相互作用能,Ecomplex为“配体/受体”复合物的能量.EL为游离配体的能量,Eb为我们定义的配体结合受体的结合能,它与结合过程中的自由能变相等,可通过Ecomplex-ER-EL计算.游离受体能量ER的计算与对接时采取的条件相同,即对配体分子12范围内的氨基酸残基不做任何限制,其余氨基酸原子的位置则保持不变,其余各项参数相同.为了比较各种能量与活性的分析效果,对所有能量都用非交叉验证PLS进行了回归分析,所得结果见表1.
, http://www.100md.com
Tab.1 The inhibitory activities and Dock energy of 12 compounds used for Dock
Compd.
Eint
Eele
Este
Ecomplex
EL
Eb
-logIC50
, http://www.100md.com
(Calcd)
-logIC50
(Exptl)
1
-50.620
-24.835
-25.785
2956.56
-12.541
-47.88
8.00
7.59
, 百拇医药
2
-51.320
-24.409
-26.911
2956.620
-9.345
-51.72
8.17
8.16
3
-52.561
-29.661
-22.900
, 百拇医药
2950.657
-16.692
-50.007
8.10
8.52
4
-59.739
-31.387
-28.352
2949.553
-15.432
-52.362
, 百拇医药
8.21
8.12
5
-53.724
-28.107
-25.617
2966.693
-16.489
-34.174
7.31
7.30
6
-55.179
, 百拇医药
-27.743
-27.436
2953.797
-12.783
-50.776
8.13
8.00
7
-33.266
-19.259
-14.007
3080.062
, http://www.100md.com
92.548
-29.8.2
7.10
6.59
8
-52.230
-21.636
-18.594
3018.333
17.124
-16.139
6.42
, http://www.100md.com 6.06
9
-41.985
-30.241
-11.744
2955.069
-37.273
-25.015
6.86
7.00
10
-51.094
-26.011
, 百拇医药
-25.083
2939.523
-48.103
-29.722
7.09
7.68
11
-52.620
-32.977
-19.643
2982.927
3.000
, 百拇医药
-37.413
7.47
7.28
12
-39.808
-27.077
-12.232
2943.743
-42.692
-30.938
7.15
7.68
, 百拇医药 ER=3017.34 kcal/mol -logIC50=5.629-0.049×Eb 〈6〉
n=12,R2=0.735,s=0.397,F=27.743
-logIC50=4.418-0.064×Eint 〈7〉
n=12,R2=0.498,s=0.522,F=9.916
-logIC50=5.140-0.088×Este 〈8〉
n=12,R2=0.248,s=0.639,F=3.291
, http://www.100md.com
-logIC50=6.039-0.068Eele 〈9〉
n=12,R2=0.339,s=0.599,F=5.127
由上述四个构效方程可知,-logIC50与Eb具有最高的回归系数,R2=0.735,因此Eb是描述COX-2选择性抑制剂结合能力的最佳参数;配体和受体相互作用能描述的是配体和受体结合的牢固程度,仅部分反映结合的潜在能力,因而-logIC50与Eint也具有一定的相关性,R=0.498;对-logIC50与Este或Eele分别进行回归,相关系数都较低,由此可初步推断选择性抑制剂并不单独以立体的或静电的相互作用与受体结合,两者是共同发挥作用的.
, http://www.100md.com
利用Dock程序模拟配体和受体之间的结合,不仅能够建立结构类型不同的抑制剂的构效方程,而且还可以确定它们的结合模式.经对接后发现,与三环系化合物相同,NS-398的环己烷环和苯环分别位于由氨基酸残基组成的疏水结合腔中,它们通过立体场与结合腔内的芳环基团相互作用.联结在苯环上的硝基则与精氨酸残基Arg120,Arg513和酪氨酸Tyr355形成一个氢键网格,NS-398和COX-2晶体对接后的位置如图4所示.与NS-398结构类似的化合物D10,D11也有相同的结合模式.与三环系化合物相比,虽然NS-398类化合物缺少联结两个苯环的中间桥连环的立体场作用,但由于硝基或者羰基氢键作用的补偿没有降低该类配体分子结合受体的能力,它们仍然具有很高的生物活性.
Fig.4 The binding model of NS-398
, http://www.100md.com
二维QSAR建立的是生物活性与物理化学参数之间的相关性,比较分子力场分析的是生物活性与场的关系,Dock操作是建立结合常数与自由能变的关系,各自的存在有其理论依据,各具特色.2D需要计算或测定物化参数,CoMFA中活性构象的确定及分子叠合规则的定义是一个难点,它们直接影响模型的精度,Dock方法是在当结合位点确定的条件下进行的,分子结构虽然差别较大,只要结合位点相同,仍能通过Dock建立构效方程.Dock在建立构效方程的优点还在于,即使结合位点不明确,Felexible Dock及Auotdock可以选择正确的结合模式,计算结合能,从而建立QSAR方程.
1国家自然科学基金资助项目 No.39770877
参 考 文 献
1.Futaki N,Takahashi S,Yokyama.NS-398,a new anti-inflammatory agent selectriely inhibitor prostagolandin G/H synthase/cycloxyfenase(COX-2) activity in vitro.prostaglandins,1994,42(1):55~59
, 百拇医药
2.Kurumbail RG,Stevens AM,Gierse JK.Structure basis for selective inhibitor of cycloxygenase-2 by anti-inflammatory agents. Nature,1996,384(19):644~648
3.Garavito RM,The cyclooxygenase-2 structure:new drugs for an old target?Nature Struct Biol,1996,(2):2637~2643
4.The program Sybyl 6.4/6.04 is available from Tripos Assoc,1699 S.Hanley Rd,St Louis, MO 63144
5.Bernstein FC,Koetzle TF,Williams GJB,et al.The X-ray structure of complexes of cycloxygensase-1 and inhibitor.J Mol Biol,1977,112(2):535~542
, http://www.100md.com
6.郭宗儒.药物化学总论.北京:中国医药科技出版社,1994.41~60
7.Picot D,Loll PJ,Garavito RM.The X-ray crystal of membral structure protein prostaglandin H2 synthase-1.Nature,1994,367(2):243~249
8.Loll P,Picot D,Garavito RM.The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase.Nature Struct Biol,1995,2(3):637~643
9.Loll P,Picot D,Garavito Rm.Synthesis and use of iodinated nonsteroidal antiinflammatory drug analogs ascrystallographic probes of the prostaglandin H2 synthase cyclooxygenase active site.Biochemistry,1996,35(23):7330~7340
收稿日期:1999-01-15, http://www.100md.com(朱七庆 屈凌波 雷新胜 郭宗儒2)
单位:中国医学科学院 中国协和医科大学药物研究所,北京 100050
关键词:环氧合酶-2;构效关系;分子图形学;对接
摘 要 环氧合酶摘 要 环氧合酶-2具有多种结构类型的抑制剂,比较分子力场分析难以建立它们的定量构效关系.根据自由能原理,抑制剂结合酶的自由能变与活性呈线性关系,本文选择了12个典型结构的的环氧合酶-2抑制剂进行对接,通过分析结合能与活性的相关性,得到了较好的构效模型,相关系数r=0.857,在建立构效模型的同时,通过图形学分析,揭示了这些抑制剂具有较高活性的原因.
Study on the Dock of Cyclooxygenase-2 Inhibitors
Zhu Qiqing,Qu Lingbo,Lei Xinsheng,Guo Zongru
, 百拇医药
Institute of Materia Medica,Chinese Academy of Medical Sciences
and Peking Union Medical College,Beijing 100050
Abstract Selective cyclooxygenase-2(COX-2) inhibitors contain varions structure skeletons.It is more difficult to construct their three dimensional quantitative structure-activity relationship by using the comparative molecular field analysis.In order to bulid their QSAR model and guide the design of a new inhibitor,in this thesis,12 selective COX-2 inhibitor were selected to dock into COX-2 enzyme by using the Dock program.The results showed that there existed a good correlation between activity-logIC50 and binding energy,the relative coefficient r=0.857.In addition Dock mimiced the binding of NS-398 analogueed to COX-2,and the results indicated that NS-398 analogues possessed higher activity because of the presence of additional hydrogen-bonding.
, 百拇医药
Key words cyclooxygenase-2;QSAR molecular graphics;Dock
研究环氧合酶-2(COX-2)特异性抑制剂,是创新新型抗炎药物的重要内容.对于三环类抑制剂,通过比较分子力场分析,已经建立了有效的三维定量构效关系模型.然而,在已有的环氧合酶抑制剂中,除了三环类化合物外,双环类化合物尼美舒利(Nimisulide)和NS-398〔1〕等也是典型的环氧合酶-2抑制剂,这些化合物结构差别较大,分子中起主要或次要作用的力场在三维空间中有可能发生位置上的改变.对于整个分子而言,由于空间性质上的涨落现象,这种场的变化未必会造成分子活性的降低,但是采用原有的叠合方式进行力场比较已经失去意义,使用CoMFA,回归力场与活性的相关性分析往往难以得到有效的QSAR方程.
由于环氧合酶-2的晶体结构已经确定,并已知与抑制剂的结合位点〔2,3〕,本文利用对接程序(Dock),通过计算系列抑制剂与环氧合酶-2的结合能,建立结构变化较大的抑制剂与活性之间的相关方程,同时探求不同结构类型抑制剂结合环氧合酶-2的特征,为环氧合酶-2抑制剂的设计提供有关结构信息.
, 百拇医药
所有计算工作都是在中国医学科学院 中国协和医科大学药物研究所合成室工作站Silicon Graphics IRIS Indigo R40000计算机工作站上完成,应用程序为Tripos公司最新提供的SYBYL 6.4分子软件包〔4〕。计算过程中除特别指明外,所选用的参数均为缺省值,环氧合酶-2与抑制剂的复合物晶体结构从PDB数据库中获得〔5〕.
1 计算方法
1.1 基本原理
药物分子发挥药效必须结合受体形成复合物,生物效应经构成复合物的药物的内在活性引发,药物的作用强度与形成复合物的数目相关,复合物的数目越多,药物的药理作用就越强〔6〕.该过程可简单描述为:]
, 百拇医药
Fig.1 The compounds used for Dock analysis
1.2 对接化合物的选择
进行对接研究的12个活性已知但结构不同的化合物结构如图1所示.这12个化合物根据亚结构特征分属于三种不同的结构类型,即三环类、双环类和吲哚美辛.考虑到对接分子的多样性,虽然吲哚美辛对于环氧合酶-2的选择性不高,但其结合COX-2的能力较高并且结构变化更大,因此选其为对接分子.所有12个化合物的活性都具有相同的测试标准,活性之间具有可参比性,活性IC50值列于图1中.
三环类的NC-398和吲哚美辛结合受体的晶体结构的解析为该类化合物的活性构象确立了实验基础(2,7~9).NC-398类化合物虽然与三环系化合物的结构有所区别,但是,药效团的分布是相似的,本文通过代表性的抑制剂NS-398的有限制条件的构象搜索,来寻找该类抑制剂的活性构象.
, http://www.100md.com
1.3 NS-398类化合物的构象分析
NS-398类化合物与三环系化合物的最大区别在于缺少联结两个芳环的中间环,醚键相连的两个苯环与砜基则与三环系中的两个芳环和砜基相对应.显然,NS-398中的这些基团在三维空间的配置应与三环系化合物一致,这两类化合物理应有相同的结合位点.如图2所示,以SC-558为例,定义该类抑制剂的药效团为砜基的硫原子(C)以及两个苯环中心(A)和(B),三个药效团原子间的距离分别表示为d1,d\-2,d\-3,在SC-558活性构象中,d1,d2,d3分别为6.993
, 百拇医药
Fig.2 Distance constraints used for conformational search
1.4 已知化合物的对接及构效方程的建立
对接操作的关键步骤是确定配体分子的初始位置.三类已知抑制剂中,三环类抑制剂的定位以复合物中配体分子SC-558为模板进行分子叠合定位,吲哚美辛的晶体结构则是已知的,经过加氢、荷载电荷直接可以进行对接.NS-398类化合物的定位则通过它的活性构象与三环系的分子叠合来进行.对接过程中选用Tripos力场优化配体和受体之间的相互作用,COX-2荷载Kollman all-atom电荷,配体分子带PM3计算的MOPAC电荷,介电常数与距离相关ε=4rij,能量收敛限为0.1 kcal/mol,计算仅考虑配体分子4
, 百拇医药
2 结果与讨论
以d1作为限制条件,经系统搜索得到10个NS-398的低能构象,这10个构象两两互成镜像,镜像分子具有相同的能量值.结构优化后,属于镜像同侧的5个构象体都属于相同的几何构型,因此,最终仅得2个分子构象,分别记为构象a和构象b,构象a中环己烷环朝内,构象b的环己烷环朝外.这两个构象与能量最低构象几乎具有相同的几何形状,主要区别在于磺酰胺侧链的方向不同,其能量差小于3.0 kcal/mol.
Fig.3 Superimposition of Conformer a,b on SC-558
根据构象分析结果,认为NS-398类化合物应以构象a或构象b与COX-2结合.由于三环系分子的活性构象是已知的,NS-398类与三环系化合物具有相同的药效团,因此通过构象a和构象b与三环系化合物SC-558活性构象的叠合,可确定NS-398的活性构象,两种构象叠合后如图4所示,构象a的环己烷环与SC-558的另一个苯环重叠,构象b的环己烷却远离苯环,由叠合图可知,构象a与SC-558活性构象具有更好的相似性,构象a可以作为NS-398类抑制剂的活性构象,以该叠合后的位置作为对接NS-398类抑制剂对接时的初始位置,构象a中距离d-1,d-2,d-3分别为6.92
, 百拇医药
表1列出了12个化合物对接后的能量项,Eint为对接后配体和受体的相互作用能,Este和Eele分别为配体和受体相互作用能中的立体相互作用能和静电相互作用能,Ecomplex为“配体/受体”复合物的能量.EL为游离配体的能量,Eb为我们定义的配体结合受体的结合能,它与结合过程中的自由能变相等,可通过Ecomplex-ER-EL计算.游离受体能量ER的计算与对接时采取的条件相同,即对配体分子12
, http://www.100md.com
Tab.1 The inhibitory activities and Dock energy of 12 compounds used for Dock
Compd.
Eint
Eele
Este
Ecomplex
EL
Eb
-logIC50
, http://www.100md.com
(Calcd)
-logIC50
(Exptl)
1
-50.620
-24.835
-25.785
2956.56
-12.541
-47.88
8.00
7.59
, 百拇医药
2
-51.320
-24.409
-26.911
2956.620
-9.345
-51.72
8.17
8.16
3
-52.561
-29.661
-22.900
, 百拇医药
2950.657
-16.692
-50.007
8.10
8.52
4
-59.739
-31.387
-28.352
2949.553
-15.432
-52.362
, 百拇医药
8.21
8.12
5
-53.724
-28.107
-25.617
2966.693
-16.489
-34.174
7.31
7.30
6
-55.179
, 百拇医药
-27.743
-27.436
2953.797
-12.783
-50.776
8.13
8.00
7
-33.266
-19.259
-14.007
3080.062
, http://www.100md.com
92.548
-29.8.2
7.10
6.59
8
-52.230
-21.636
-18.594
3018.333
17.124
-16.139
6.42
, http://www.100md.com 6.06
9
-41.985
-30.241
-11.744
2955.069
-37.273
-25.015
6.86
7.00
10
-51.094
-26.011
, 百拇医药
-25.083
2939.523
-48.103
-29.722
7.09
7.68
11
-52.620
-32.977
-19.643
2982.927
3.000
, 百拇医药
-37.413
7.47
7.28
12
-39.808
-27.077
-12.232
2943.743
-42.692
-30.938
7.15
7.68
, 百拇医药 ER=3017.34 kcal/mol -logIC50=5.629-0.049×Eb 〈6〉
n=12,R2=0.735,s=0.397,F=27.743
-logIC50=4.418-0.064×Eint 〈7〉
n=12,R2=0.498,s=0.522,F=9.916
-logIC50=5.140-0.088×Este 〈8〉
n=12,R2=0.248,s=0.639,F=3.291
, http://www.100md.com
-logIC50=6.039-0.068Eele 〈9〉
n=12,R2=0.339,s=0.599,F=5.127
由上述四个构效方程可知,-logIC50与Eb具有最高的回归系数,R2=0.735,因此Eb是描述COX-2选择性抑制剂结合能力的最佳参数;配体和受体相互作用能描述的是配体和受体结合的牢固程度,仅部分反映结合的潜在能力,因而-logIC50与Eint也具有一定的相关性,R=0.498;对-logIC50与Este或Eele分别进行回归,相关系数都较低,由此可初步推断选择性抑制剂并不单独以立体的或静电的相互作用与受体结合,两者是共同发挥作用的.
, http://www.100md.com
利用Dock程序模拟配体和受体之间的结合,不仅能够建立结构类型不同的抑制剂的构效方程,而且还可以确定它们的结合模式.经对接后发现,与三环系化合物相同,NS-398的环己烷环和苯环分别位于由氨基酸残基组成的疏水结合腔中,它们通过立体场与结合腔内的芳环基团相互作用.联结在苯环上的硝基则与精氨酸残基Arg120,Arg513和酪氨酸Tyr355形成一个氢键网格,NS-398和COX-2晶体对接后的位置如图4所示.与NS-398结构类似的化合物D10,D11也有相同的结合模式.与三环系化合物相比,虽然NS-398类化合物缺少联结两个苯环的中间桥连环的立体场作用,但由于硝基或者羰基氢键作用的补偿没有降低该类配体分子结合受体的能力,它们仍然具有很高的生物活性.
Fig.4 The binding model of NS-398
, http://www.100md.com
二维QSAR建立的是生物活性与物理化学参数之间的相关性,比较分子力场分析的是生物活性与场的关系,Dock操作是建立结合常数与自由能变的关系,各自的存在有其理论依据,各具特色.2D需要计算或测定物化参数,CoMFA中活性构象的确定及分子叠合规则的定义是一个难点,它们直接影响模型的精度,Dock方法是在当结合位点确定的条件下进行的,分子结构虽然差别较大,只要结合位点相同,仍能通过Dock建立构效方程.Dock在建立构效方程的优点还在于,即使结合位点不明确,Felexible Dock及Auotdock可以选择正确的结合模式,计算结合能,从而建立QSAR方程.
1国家自然科学基金资助项目 No.39770877
参 考 文 献
1.Futaki N,Takahashi S,Yokyama.NS-398,a new anti-inflammatory agent selectriely inhibitor prostagolandin G/H synthase/cycloxyfenase(COX-2) activity in vitro.prostaglandins,1994,42(1):55~59
, 百拇医药
2.Kurumbail RG,Stevens AM,Gierse JK.Structure basis for selective inhibitor of cycloxygenase-2 by anti-inflammatory agents. Nature,1996,384(19):644~648
3.Garavito RM,The cyclooxygenase-2 structure:new drugs for an old target?Nature Struct Biol,1996,(2):2637~2643
4.The program Sybyl 6.4/6.04 is available from Tripos Assoc,1699 S.Hanley Rd,St Louis, MO 63144
5.Bernstein FC,Koetzle TF,Williams GJB,et al.The X-ray structure of complexes of cycloxygensase-1 and inhibitor.J Mol Biol,1977,112(2):535~542
, http://www.100md.com
6.郭宗儒.药物化学总论.北京:中国医药科技出版社,1994.41~60
7.Picot D,Loll PJ,Garavito RM.The X-ray crystal of membral structure protein prostaglandin H2 synthase-1.Nature,1994,367(2):243~249
8.Loll P,Picot D,Garavito RM.The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase.Nature Struct Biol,1995,2(3):637~643
9.Loll P,Picot D,Garavito Rm.Synthesis and use of iodinated nonsteroidal antiinflammatory drug analogs ascrystallographic probes of the prostaglandin H2 synthase cyclooxygenase active site.Biochemistry,1996,35(23):7330~7340
收稿日期:1999-01-15, http://www.100md.com(朱七庆 屈凌波 雷新胜 郭宗儒2)