环氧合酶结合抑制剂的分子图形学研究1
作者:朱七庆 屈凌波 雷新胜 郭宗儒
单位:中国医学科学院中国协和医科大学药物研究所,北京100050
关键词:环氧合酶-1;环氧合酶-2;分子图形学;对接
摘 要 环氧合酶具有两种结构亚型摘 要 环氧合酶具有两种结构亚型,即环氧合酶-1和环氧合酶-2,环氧合酶-2选择性抑制剂的毒副作用低,是较好的非甾体抗炎药物.本文通过选择性抑制剂SC-558和非选择性抑制剂吲哚美辛分别与环氧合酶-1和-2进行对接,利用分子图形学,从定量和定性的角度阐明了抑制剂选择结合环氧合酶-1和-2的原因.
Molecular Graphics Study on the Binding of
Cyclooxygenase to Their Inhibitors
, 百拇医药
Zhu Qiqing,Qu Lingbo,Lei Xinsheng,Guo Zhongru
(Institute of Materia Medica,Chinese Academy of Medical Sciences
and Peking Union Medical College,Beijing 100050)
Abstract The cyclooxygenase(COX)exists in two subtypes,a cons titutive form of cyclooxygenase-1(COX-1)and an inducible form of cyclooxygenas e-2( COX-2).Selective inhibitors of COX-2 are effective antiinflammatory agents w i thout side-effects associated with current NSAIDs(non steroidal antiinflammator y drugs).In order to understand the selectivity of cyclooxygenase-2 inhibitors, a selective COX-2 inhibitor SC-558 and a non-selective inhibitor indomethacin w e re docked into COX-1 and COX-2,respectively.The results from molecular graphic s visualized the binding feature and explained their selectivity.
, 百拇医药
Key words cyclooxygenase-1;cyclooxygenase-2;molecular graph ics;Dock
选择性环氧合酶-2(COX-2)抑制剂仅阻断炎症介质前列腺素的生成,因而与经典的非甾体抗炎药(NSAIDs)不同,它产生的毒、副作用较小,能够避免胃肠道和肾脏的损伤〔1〕.环氧合酶两种结构亚型环氧合酶-1(COX-1)和环氧合酶-2(COX-2)分别与抑制剂结合形成的复合物晶体结构已被解析〔2,3〕,通过分析抑制剂在结合腔中与酶的相互作用特征,可以确定酶与抑制剂结合的药效团分布,从而定向设计具有高选择性的环氧合酶-2抑制剂.然而,环氧合酶-1和环氧合酶-2具有较高的同源性(约67%),它们的活性中心具有类似拓扑结构,此外,环氧化活性中心起主要作用的Tyr385,Arg120等氨基酸都是保守的,两种酶的重要区别仅在于523位氨基酸,环氧合酶-1在该位置为异亮氨酸Ile523,环氧合酶-2则为丙氨酸Ala523.从环氧合酶-1和环氧合酶-2的晶体结构难以直接发现它们选择结合抑制剂的原因.基于X-ray晶体结构的结果,本文通过选择性和非选择性环氧合酶-2抑制剂分别对这两种酶进行对接(Dock)研究,从对接后形成“复合物”的能量变化以及具体的结合模式等多个方面探讨环氧合酶选择结合抑制剂的内在原因.
, http://www.100md.com
1 计算方法
所有计算工作都是在中国医学科学院、中国协和医科大学药物研究所合成室的SGIIndigoR4000计算机工作站上完成的,所用计算程序为Tripos公司最新提供的Sybyl6.4〔4〕分子设计软件包.计算过程中除非特别指明,所选用的参数均为缺省值.环氧合酶-1和环氧合酶-2结合抑制剂的复合物晶体结构数据从PDB数据库中获得〔5〕.
1.1 对接分子的选择
SC-558是一个选择性环氧合酶-2抑制剂,对环氧合酶-1和-2的半数抑制浓度IC50分别为17.7 μmol/L和0.0093μmol/L,选择性高达1900倍〔6〕.SC-558结合环氧合酶-2的晶体结构虽然已被解析〔7〕,但由于它与环氧合酶-1的结合能力低,相应的晶体结构难以获得,因而与环氧合酶-1的相互作用只能通过对接的方法加以模拟.吲哚美辛抑制环氧合酶-1和-2的IC50分别为0.1μmol/L和0.9μmol/L〔6〕,它们的复合物晶体结构也已被解析〔8,9〕.两个用于对接研究的抑制剂分子SC-558和吲哚美辛如图1所示.
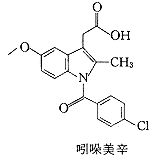
, http://www.100md.com
Fig.1 The compounds used for Dock to COX-1 and COX-2
1.2 对接分子的预处理
环氧合酶-1和-2的晶体结构以PDB格式存贮,对接之前需经分子力学进行几何构型优化.由于与抑制剂分子直接或间接发生相互作用的仅是其附近一定范围内的氨基酸残基,因此在进行构型优化时,仅对抑制剂分子12?范围内的氨基酸残基进行能量优化,其余原子的构型在整个优化过程中始终保持不变.能量优化用分子力学程序MINIMIZE2,力场为Tripos标准力场,蛋白分子带Kollmanall-atom电荷,能量收敛限为0.1kcal/mol,介电常数与距离相关,ε=4rij,能量收敛后,环氧合酶-1和环氧合酶-2的三维结构均没有发生明显的变化.对于抑制剂分子SC-558和吲哚美辛也进行几何构型优化,与酶分子相比,所不同的只是计算过程中抑制剂分子带MOPAC电荷,能量收敛限为0.05kcal/mol,介电常数ε=rij.
, 百拇医药
1.3 分子的对接
在进行对接前,必须确定酶和抑制剂的初始位置.对于SC-558与环氧合酶-2的对接,定位原则是以复合物晶体结构为模板,将优化所得环氧合酶-2及SC-558与之叠合,环氧合酶-2以几何构型保持不变的氨基酸残基主肽链原子为叠合原子,SC-558的叠合原子为三个环的中心.在SC-558与环氧合酶-1的对接中,环氧合酶-1和SC-558的定位方法与上相同.对接过程中能量优化所选各项参数:力场为Tripos标准力场,蛋白分子带Kollmanall-atom电荷,抑制剂分子带MOPAC电荷,能量收敛限为0.1kcal/mol,介电常数与距离相关,ε=4rij.
非选择性抑制剂吲哚美辛与环氧合酶-1和-2的对接方法与SC-558相同.
2 结果与讨论
2.1 选择性抑制剂SC-558与环氧合酶-1和环氧合酶-2的对接结果
, http://www.100md.com
SC-558对接环氧合酶-1和-2后的各能量项及氢键原子间的距离列于表1中,d1为磺酰氨基的氢原子到Phe513的羰基氧的距离,d2和d3分别为两个氧原子到His90和Arg513羟基氢原子的距离,这些距离的大小表明形成氢键能力的强弱.在忽略熵变的情况下,对接后系统自由能变ΔG=Ecomplex-ER-EL,Ecomplex为复合物的能量,ER为游离酶的能量,EL为活性构象是抑制剂分子的能量,以此计算的自由能变能够反映抑制剂和酶之间结合的难易程度.表中的数据提示,SC-558结合环氧合酶-2的自由能变小于结合环氧合酶-1的自由能变,即ΔGcox-2=-50.4kcal/mol<ΔGcox-1=-11.4kcal/mol;自由能变数值越小,表明抑制剂和酶就越容易结合,形成的复合物越稳定.因此,从选择性抑制剂SC-558与两种环氧合酶结合的自由能变,定量地支持了该类抑制剂优先选择结合环氧合酶-2.
, http://www.100md.com
Tab.1 Energy and distance changes after Dock of SC -558
Enzyme
ΔER
(kcal/mol)
ΔEL
(kcal/mol)
ΔG
(kcal/m ol)
d1
( )
)
, http://www.100md.com
d2
()
d3
()
COX-1
53.3
21.13
-11.4
, 百拇医药
4.47
3 .86
4.02
COX-2
6.77
4.13
-50.4
2.18
2.89
2.46
SC-558类化合物的选择性从它与两种同功酶的结合模式中也可得到解释,图2显示了对接后受体和配体的相互作用.在环氧合酶-2中,523位丙氨酸氨基酸残基周围存在两个腔穴,分别容纳SC-558的溴代苯环和磺酰胺基苯环,整个分子以一个叉形的结构与环氧合酶-2结合.溴代苯环与周围的Phe381,Leu384,Tyr385,Trp387,Phe513和Ser530以及Gly526和Ala527相互作用;三氟甲基吡唑环则与由氨基酸Val116,Val349,Tyr355,Leu359,Leu531形成的疏水腔发生相互作用;磺酰胺代苯环则结合于由Leu352,Tyr355,Phe518,Val523以及Ser353的主肽链原子形成的疏水腔中.SC-558的磺酰胺基延伸到环氧合酶-2的表面,与一些极性的氨基酸残基相互作用,如磺酰胺基的一个氧原子与组氨酸His90形成氢键,另一个氧原子则与精氨酸Arg513形成另一个氢键,氨基的氮原子与其附近的Phe513氨基酸形成氢键,所有这些相互作用对于SC-558以高亲和力结合环氧合酶-2都是必需的.SC-558对接环氧合酶-2后,酶和抑制剂的能量变化均小于7.0kcal/mol,这从另一个方面说明SC-558与环氧合酶-2结合是合理的,该类结合方式并没有造成不合理的能量变化,它们之间不存在大的张力.从形成氢键原子间的距离d1=2.18,d2=2.89和d3=2.46也可以看出,抑制剂和酶之间形成较强的氢键.SC-558与环氧合酶-1对接后,能量都有较大的升高,从抑制剂分子SC-558来看,该分子发生了严重的扭曲,能量升高了15.0kcal/mol,SC-558的磺酰基没有充分地延伸到分子的表面并与相应的极性基团发生氢键作用,相应原子之间的距离d1=4.47,d2=3.86和d3=4.02也说明了抑制剂SC-558与环氧合酶-1之间的氢键作用甚弱,结合不利.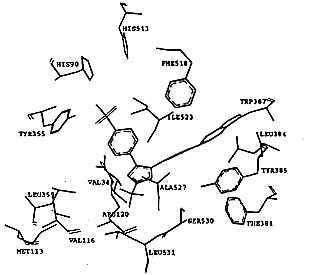

, http://www.100md.com
Fig.2 The model of Dock of SC-558 into COX-1 and COX-2
进一步分析可知,造成这一结果的主要原因在于结合腔中523位氨基酸残基的不同,在环氧合酶-2中,523位为缬氨酸Val523,它与其它氨基酸残基Tyr355,Leu352,Ser353,Phe518形成体积较大的腔穴,能够容纳磺酰胺代苯环的进入并与其发生相互作用.在环氧合酶-1中,523位为异亮氨酸Ile523,由于Ile的体积大于Val,因此在环氧合酶-1中,由Ile523,Tyr355,Leu352,Ser353,Phe518形成的结合腔体积比环氧合酶-2的小,结合腔体积的缩小,使得抑制剂分子不易进入该腔穴,对接的结果说明了抑制剂进入这一腔穴是比较困难的,这也进一步说明了SC-558选择结合环氧合酶-2的原因,对于与SC-558相类似的三环系化合物,其选择性都可以从SC-558的结论中得到阐明.
2.2 非选择性抑制剂吲哚美辛与环氧合酶-1和环氧合酶-2的对接结果
, 百拇医药
非选择性抑制剂吲哚美辛对接环氧合酶-1和环氧合酶-2后的各能量项见表2.由表中数据可知,吲哚美辛结合环氧合酶-2的自由能变ΔGcox-2=-32.80kcal/mol与结合环氧合酶-1的自由能变ΔGcox-1=-25.75kcal/mol相差不大,这表明吲哚美辛对COX-1和COX-2具有相近的亲和力.因而,吲哚美辛非选择地结合于这两种同功酶.从对接后吲哚美辛和酶的能量变化来考察,其结合也都是合理的,结合后吲哚美辛和酶的能量变化均未超过8.0kcal/mol.
Tab.2 The energy changes of indomethacin Dock into COX
kcal/mol
Enzyme
ΔER
, 百拇医药
ΔEL
ΔG
COX-1
6.77
4.82
-32.80
COX-2
7.89
5.37
-25.75
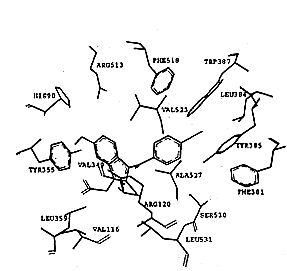
吲哚美辛与两种同功酶的非选择性抑制主要原因在于它的结合位点,如图3所示.在环氧合酶-2-吲哚美辛“复合物”中,吲哚美辛采取cis-构型与环氧合酶结合,羧基与精氨酸Arg120形成盐桥键,吲哚环与缬氨酸Val349和丝氨酸Ser353相互作用,六元环则接近Leu352和Ser353,吲哚环上的甲氧基深入到Ser353,Tyr355和Val523所组成的结合腔中,氯苯基环相当于SC-558的溴苯基,而吡咯环相当于SC-558中的吡唑环,它们结合于相同的结合腔中.在环氧合酶-1-吲哚美辛“复合物”中,吲哚美辛与环氧合酶-1的作用跟环氧合酶-2非常相似,明显区别只是在苯甲酰基的羰基氧原子上,在环氧合酶-1中,苯甲酰基的羰基氧原子与Ser530的羟基形成氢键,而在环氧合酶-2中没有发现这两个基团之间存在氢键相互作用,吲哚美辛抑制环氧合酶-1的IC50(0.08μmol/L)略高于抑制环氧合酶-2的IC50(0.96μmol/L),也许是由于在环氧合酶-1复合物中存在这一氢键相互作用.吲哚美辛的非选择性结合主要原因是它远离由Leu352, Tyr355, Phe518, Val523或Ile523以及Ser353形成的疏水腔,不能直接与该腔穴的氨基酸残基发生作用,因此,当对抑制剂选择性起主要作用的Val523氨基酸残基被COX-1的异亮氨酸Ile取代后,并没有对吲哚美辛的结合产生大的影响,这是吲哚美辛缺乏选择性的原因所在.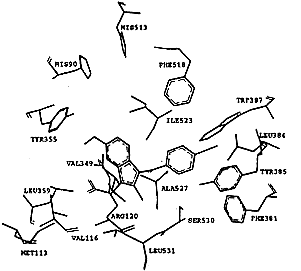
, http://www.100md.com
Fig.3 The model of Dock of indomethacin into COX-1 and COX-2
以上研究表明,自由能变ΔG是衡量配体与受体相互结合的定量指标,通过对接计算的自由能变与抑制剂和酶相互结合的难易程度是一致的.借助于分子图形学,还对环氧合酶-1和环氧合酶-2结合选择性抑制剂和非选择性抑制剂的结合模式进行了研究.所得结果表明:造成抑制剂选择性结合两种同功酶的原因是它们的结合位点不同,选择性抑制剂结合的腔穴对于环氧合酶-1和环氧合酶-2有较大的区别,非选择性抑制剂结合则具有较大的相似性.
1国家自然科学基金资助项目No.39770877
郭宗儒:通讯联系人
参考文献
1 Xie W,Robertson DL,Simmons DL.Mitogen inducible prostaglandin G/H syn thesis:a new target for nonsteroidal antiinflammatory drugs.Drug Dev Res,1992,25 (2):249~265
, 百拇医药
2 Picot D,Loll PJ,Garavito RM.The X-ray crystal of membral structure protein p rostaglandin H2 synthase-1.Nature,1994,367(20):243~249
3 Luong C,Miller A,Barnett J,et al.Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2.Nature Structural Biology,1996,3(11):9 27~933
4 The program Sybyl 6.4/6.04 is available from Tripos Assoc,1699 S.Hanley Rd, St Louis,MO 63144
, 百拇医药
5 Bernstein FC,Koetzle TF,Williams GJB,et al.The protein databank:computer -base archiral file for marcromolecular structures.J Mol Biol,1977,112(3):535 ~542
6 Gierse JK, Hauser SD,Isakson PC,et al.Expression and selective inhibition of the constitutive and inducible forms of human cyclo-oxygenase.Biochemistry J,1995,305(2):479~48 4
7 Kurumbail RG,Stevens AM,Gierse JK.Structure basis for selective inhibitor of cycloxygenase-2 by antiinflammatory agents.Nature,1996,384(19/26):644~648
, 百拇医药
8 Loll PJ,Picot D,Ekabo O,et al.Synthesis and use of iodinated nonsteron an t iinflammatory drug analogs as crystallographic probes of the prostaglandin H syn thase cyclooxygenase active site.Biochemistry,1996,35(23):7330~7340
9 Black WC,Bayly C,Prasit P,et al.Fromindomethacin to a selective COX-2 in hibitor:developm ent of indolakanoic acid as potent and selective cyclooxygenase-2 inhibitor.Bio org Med Chem Lett,1996,6(6):725~730
收稿日期:1999-01-18, http://www.100md.com
单位:中国医学科学院中国协和医科大学药物研究所,北京100050
关键词:环氧合酶-1;环氧合酶-2;分子图形学;对接
摘 要 环氧合酶具有两种结构亚型摘 要 环氧合酶具有两种结构亚型,即环氧合酶-1和环氧合酶-2,环氧合酶-2选择性抑制剂的毒副作用低,是较好的非甾体抗炎药物.本文通过选择性抑制剂SC-558和非选择性抑制剂吲哚美辛分别与环氧合酶-1和-2进行对接,利用分子图形学,从定量和定性的角度阐明了抑制剂选择结合环氧合酶-1和-2的原因.
Molecular Graphics Study on the Binding of
Cyclooxygenase to Their Inhibitors
, 百拇医药
Zhu Qiqing,Qu Lingbo,Lei Xinsheng,Guo Zhongru
(Institute of Materia Medica,Chinese Academy of Medical Sciences
and Peking Union Medical College,Beijing 100050)
Abstract The cyclooxygenase(COX)exists in two subtypes,a cons titutive form of cyclooxygenase-1(COX-1)and an inducible form of cyclooxygenas e-2( COX-2).Selective inhibitors of COX-2 are effective antiinflammatory agents w i thout side-effects associated with current NSAIDs(non steroidal antiinflammator y drugs).In order to understand the selectivity of cyclooxygenase-2 inhibitors, a selective COX-2 inhibitor SC-558 and a non-selective inhibitor indomethacin w e re docked into COX-1 and COX-2,respectively.The results from molecular graphic s visualized the binding feature and explained their selectivity.
, 百拇医药
Key words cyclooxygenase-1;cyclooxygenase-2;molecular graph ics;Dock
选择性环氧合酶-2(COX-2)抑制剂仅阻断炎症介质前列腺素的生成,因而与经典的非甾体抗炎药(NSAIDs)不同,它产生的毒、副作用较小,能够避免胃肠道和肾脏的损伤〔1〕.环氧合酶两种结构亚型环氧合酶-1(COX-1)和环氧合酶-2(COX-2)分别与抑制剂结合形成的复合物晶体结构已被解析〔2,3〕,通过分析抑制剂在结合腔中与酶的相互作用特征,可以确定酶与抑制剂结合的药效团分布,从而定向设计具有高选择性的环氧合酶-2抑制剂.然而,环氧合酶-1和环氧合酶-2具有较高的同源性(约67%),它们的活性中心具有类似拓扑结构,此外,环氧化活性中心起主要作用的Tyr385,Arg120等氨基酸都是保守的,两种酶的重要区别仅在于523位氨基酸,环氧合酶-1在该位置为异亮氨酸Ile523,环氧合酶-2则为丙氨酸Ala523.从环氧合酶-1和环氧合酶-2的晶体结构难以直接发现它们选择结合抑制剂的原因.基于X-ray晶体结构的结果,本文通过选择性和非选择性环氧合酶-2抑制剂分别对这两种酶进行对接(Dock)研究,从对接后形成“复合物”的能量变化以及具体的结合模式等多个方面探讨环氧合酶选择结合抑制剂的内在原因.
, http://www.100md.com
1 计算方法
所有计算工作都是在中国医学科学院、中国协和医科大学药物研究所合成室的SGIIndigoR4000计算机工作站上完成的,所用计算程序为Tripos公司最新提供的Sybyl6.4〔4〕分子设计软件包.计算过程中除非特别指明,所选用的参数均为缺省值.环氧合酶-1和环氧合酶-2结合抑制剂的复合物晶体结构数据从PDB数据库中获得〔5〕.
1.1 对接分子的选择
SC-558是一个选择性环氧合酶-2抑制剂,对环氧合酶-1和-2的半数抑制浓度IC50分别为17.7 μmol/L和0.0093μmol/L,选择性高达1900倍〔6〕.SC-558结合环氧合酶-2的晶体结构虽然已被解析〔7〕,但由于它与环氧合酶-1的结合能力低,相应的晶体结构难以获得,因而与环氧合酶-1的相互作用只能通过对接的方法加以模拟.吲哚美辛抑制环氧合酶-1和-2的IC50分别为0.1μmol/L和0.9μmol/L〔6〕,它们的复合物晶体结构也已被解析〔8,9〕.两个用于对接研究的抑制剂分子SC-558和吲哚美辛如图1所示.
, http://www.100md.com
Fig.1 The compounds used for Dock to COX-1 and COX-2
1.2 对接分子的预处理
环氧合酶-1和-2的晶体结构以PDB格式存贮,对接之前需经分子力学进行几何构型优化.由于与抑制剂分子直接或间接发生相互作用的仅是其附近一定范围内的氨基酸残基,因此在进行构型优化时,仅对抑制剂分子12?范围内的氨基酸残基进行能量优化,其余原子的构型在整个优化过程中始终保持不变.能量优化用分子力学程序MINIMIZE2,力场为Tripos标准力场,蛋白分子带Kollmanall-atom电荷,能量收敛限为0.1kcal/mol,介电常数与距离相关,ε=4rij,能量收敛后,环氧合酶-1和环氧合酶-2的三维结构均没有发生明显的变化.对于抑制剂分子SC-558和吲哚美辛也进行几何构型优化,与酶分子相比,所不同的只是计算过程中抑制剂分子带MOPAC电荷,能量收敛限为0.05kcal/mol,介电常数ε=rij.
, 百拇医药
1.3 分子的对接
在进行对接前,必须确定酶和抑制剂的初始位置.对于SC-558与环氧合酶-2的对接,定位原则是以复合物晶体结构为模板,将优化所得环氧合酶-2及SC-558与之叠合,环氧合酶-2以几何构型保持不变的氨基酸残基主肽链原子为叠合原子,SC-558的叠合原子为三个环的中心.在SC-558与环氧合酶-1的对接中,环氧合酶-1和SC-558的定位方法与上相同.对接过程中能量优化所选各项参数:力场为Tripos标准力场,蛋白分子带Kollmanall-atom电荷,抑制剂分子带MOPAC电荷,能量收敛限为0.1kcal/mol,介电常数与距离相关,ε=4rij.
非选择性抑制剂吲哚美辛与环氧合酶-1和-2的对接方法与SC-558相同.
2 结果与讨论
2.1 选择性抑制剂SC-558与环氧合酶-1和环氧合酶-2的对接结果
, http://www.100md.com
SC-558对接环氧合酶-1和-2后的各能量项及氢键原子间的距离列于表1中,d1为磺酰氨基的氢原子到Phe513的羰基氧的距离,d2和d3分别为两个氧原子到His90和Arg513羟基氢原子的距离,这些距离的大小表明形成氢键能力的强弱.在忽略熵变的情况下,对接后系统自由能变ΔG=Ecomplex-ER-EL,Ecomplex为复合物的能量,ER为游离酶的能量,EL为活性构象是抑制剂分子的能量,以此计算的自由能变能够反映抑制剂和酶之间结合的难易程度.表中的数据提示,SC-558结合环氧合酶-2的自由能变小于结合环氧合酶-1的自由能变,即ΔGcox-2=-50.4kcal/mol<ΔGcox-1=-11.4kcal/mol;自由能变数值越小,表明抑制剂和酶就越容易结合,形成的复合物越稳定.因此,从选择性抑制剂SC-558与两种环氧合酶结合的自由能变,定量地支持了该类抑制剂优先选择结合环氧合酶-2.
, http://www.100md.com
Tab.1 Energy and distance changes after Dock of SC -558
Enzyme
ΔER
(kcal/mol)
ΔEL
(kcal/mol)
ΔG
(kcal/m ol)
d1
(
, http://www.100md.com
d2
(
d3
(
COX-1
53.3
21.13
-11.4
, 百拇医药
4.47
3 .86
4.02
COX-2
6.77
4.13
-50.4
2.18
2.89
2.46
SC-558类化合物的选择性从它与两种同功酶的结合模式中也可得到解释,图2显示了对接后受体和配体的相互作用.在环氧合酶-2中,523位丙氨酸氨基酸残基周围存在两个腔穴,分别容纳SC-558的溴代苯环和磺酰胺基苯环,整个分子以一个叉形的结构与环氧合酶-2结合.溴代苯环与周围的Phe381,Leu384,Tyr385,Trp387,Phe513和Ser530以及Gly526和Ala527相互作用;三氟甲基吡唑环则与由氨基酸Val116,Val349,Tyr355,Leu359,Leu531形成的疏水腔发生相互作用;磺酰胺代苯环则结合于由Leu352,Tyr355,Phe518,Val523以及Ser353的主肽链原子形成的疏水腔中.SC-558的磺酰胺基延伸到环氧合酶-2的表面,与一些极性的氨基酸残基相互作用,如磺酰胺基的一个氧原子与组氨酸His90形成氢键,另一个氧原子则与精氨酸Arg513形成另一个氢键,氨基的氮原子与其附近的Phe513氨基酸形成氢键,所有这些相互作用对于SC-558以高亲和力结合环氧合酶-2都是必需的.SC-558对接环氧合酶-2后,酶和抑制剂的能量变化均小于7.0kcal/mol,这从另一个方面说明SC-558与环氧合酶-2结合是合理的,该类结合方式并没有造成不合理的能量变化,它们之间不存在大的张力.从形成氢键原子间的距离d1=2.18
, http://www.100md.com
Fig.2 The model of Dock of SC-558 into COX-1 and COX-2
进一步分析可知,造成这一结果的主要原因在于结合腔中523位氨基酸残基的不同,在环氧合酶-2中,523位为缬氨酸Val523,它与其它氨基酸残基Tyr355,Leu352,Ser353,Phe518形成体积较大的腔穴,能够容纳磺酰胺代苯环的进入并与其发生相互作用.在环氧合酶-1中,523位为异亮氨酸Ile523,由于Ile的体积大于Val,因此在环氧合酶-1中,由Ile523,Tyr355,Leu352,Ser353,Phe518形成的结合腔体积比环氧合酶-2的小,结合腔体积的缩小,使得抑制剂分子不易进入该腔穴,对接的结果说明了抑制剂进入这一腔穴是比较困难的,这也进一步说明了SC-558选择结合环氧合酶-2的原因,对于与SC-558相类似的三环系化合物,其选择性都可以从SC-558的结论中得到阐明.
2.2 非选择性抑制剂吲哚美辛与环氧合酶-1和环氧合酶-2的对接结果
, 百拇医药
非选择性抑制剂吲哚美辛对接环氧合酶-1和环氧合酶-2后的各能量项见表2.由表中数据可知,吲哚美辛结合环氧合酶-2的自由能变ΔGcox-2=-32.80kcal/mol与结合环氧合酶-1的自由能变ΔGcox-1=-25.75kcal/mol相差不大,这表明吲哚美辛对COX-1和COX-2具有相近的亲和力.因而,吲哚美辛非选择地结合于这两种同功酶.从对接后吲哚美辛和酶的能量变化来考察,其结合也都是合理的,结合后吲哚美辛和酶的能量变化均未超过8.0kcal/mol.
Tab.2 The energy changes of indomethacin Dock into COX
kcal/mol
Enzyme
ΔER
, 百拇医药
ΔEL
ΔG
COX-1
6.77
4.82
-32.80
COX-2
7.89
5.37
-25.75
吲哚美辛与两种同功酶的非选择性抑制主要原因在于它的结合位点,如图3所示.在环氧合酶-2-吲哚美辛“复合物”中,吲哚美辛采取cis-构型与环氧合酶结合,羧基与精氨酸Arg120形成盐桥键,吲哚环与缬氨酸Val349和丝氨酸Ser353相互作用,六元环则接近Leu352和Ser353,吲哚环上的甲氧基深入到Ser353,Tyr355和Val523所组成的结合腔中,氯苯基环相当于SC-558的溴苯基,而吡咯环相当于SC-558中的吡唑环,它们结合于相同的结合腔中.在环氧合酶-1-吲哚美辛“复合物”中,吲哚美辛与环氧合酶-1的作用跟环氧合酶-2非常相似,明显区别只是在苯甲酰基的羰基氧原子上,在环氧合酶-1中,苯甲酰基的羰基氧原子与Ser530的羟基形成氢键,而在环氧合酶-2中没有发现这两个基团之间存在氢键相互作用,吲哚美辛抑制环氧合酶-1的IC50(0.08μmol/L)略高于抑制环氧合酶-2的IC50(0.96μmol/L),也许是由于在环氧合酶-1复合物中存在这一氢键相互作用.吲哚美辛的非选择性结合主要原因是它远离由Leu352, Tyr355, Phe518, Val523或Ile523以及Ser353形成的疏水腔,不能直接与该腔穴的氨基酸残基发生作用,因此,当对抑制剂选择性起主要作用的Val523氨基酸残基被COX-1的异亮氨酸Ile取代后,并没有对吲哚美辛的结合产生大的影响,这是吲哚美辛缺乏选择性的原因所在.
, http://www.100md.com
Fig.3 The model of Dock of indomethacin into COX-1 and COX-2
以上研究表明,自由能变ΔG是衡量配体与受体相互结合的定量指标,通过对接计算的自由能变与抑制剂和酶相互结合的难易程度是一致的.借助于分子图形学,还对环氧合酶-1和环氧合酶-2结合选择性抑制剂和非选择性抑制剂的结合模式进行了研究.所得结果表明:造成抑制剂选择性结合两种同功酶的原因是它们的结合位点不同,选择性抑制剂结合的腔穴对于环氧合酶-1和环氧合酶-2有较大的区别,非选择性抑制剂结合则具有较大的相似性.
1国家自然科学基金资助项目No.39770877
郭宗儒:通讯联系人
参考文献
1 Xie W,Robertson DL,Simmons DL.Mitogen inducible prostaglandin G/H syn thesis:a new target for nonsteroidal antiinflammatory drugs.Drug Dev Res,1992,25 (2):249~265
, 百拇医药
2 Picot D,Loll PJ,Garavito RM.The X-ray crystal of membral structure protein p rostaglandin H2 synthase-1.Nature,1994,367(20):243~249
3 Luong C,Miller A,Barnett J,et al.Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2.Nature Structural Biology,1996,3(11):9 27~933
4 The program Sybyl 6.4/6.04 is available from Tripos Assoc,1699 S.Hanley Rd, St Louis,MO 63144
, 百拇医药
5 Bernstein FC,Koetzle TF,Williams GJB,et al.The protein databank:computer -base archiral file for marcromolecular structures.J Mol Biol,1977,112(3):535 ~542
6 Gierse JK, Hauser SD,Isakson PC,et al.Expression and selective inhibition of the constitutive and inducible forms of human cyclo-oxygenase.Biochemistry J,1995,305(2):479~48 4
7 Kurumbail RG,Stevens AM,Gierse JK.Structure basis for selective inhibitor of cycloxygenase-2 by antiinflammatory agents.Nature,1996,384(19/26):644~648
, 百拇医药
8 Loll PJ,Picot D,Ekabo O,et al.Synthesis and use of iodinated nonsteron an t iinflammatory drug analogs as crystallographic probes of the prostaglandin H syn thase cyclooxygenase active site.Biochemistry,1996,35(23):7330~7340
9 Black WC,Bayly C,Prasit P,et al.Fromindomethacin to a selective COX-2 in hibitor:developm ent of indolakanoic acid as potent and selective cyclooxygenase-2 inhibitor.Bio org Med Chem Lett,1996,6(6):725~730
收稿日期:1999-01-18, http://www.100md.com