进行性腓骨肌萎缩症1A型研究进展
笪宇威 沈定国
关键词:进行性腓骨肌萎缩1A型 Charcot-Marie-Tooth病 遗传性运动感觉神经病
进行性腓骨肌萎缩症(Charcot-Marie-Tooth病,CMT),亦称为遗传性运动感觉神经病(hereditary motor and sen-sory neuropathy,HMSN),是神经系统常见的遗传性疾病之一,国外文献报道其发病率在1/2500~1/10000之间[1,2]。其中CMT1是最常见的亚型,其临床特点为:对称性肢体远端肌肉萎缩,慢性进行性力弱及弓形足畸形;电生理特征为运动感觉神经传导速度下降;组织化学可见髓神经纤维的数量减少,节段性脱髓鞘和髓鞘再生,形成典型的“洋葱球”结构。连锁分析证明CMT1是遗传异质性疾病,至少有4个位点。常染色体显性遗传的CMT1A、CMT1B和X连锁的显性CMTX。他们分别定位于17p11.2、1q21.3-23和Xq13,此外,CMT1C尚未定位。CMT1A是CMT1中最常见的类型,也是近年来研究的热点,我们就其分子生物学研究进展及临床应用情况综述如下。
, http://www.100md.com
1 基因序列重复片段的发现
1989年,Raeymaekers等[3]首次通过连锁分析将CMT1A的基因定位于17p11.2。随后于1991年Lupski等[4]发现在6个大French-AcadianCMT1A家系和1个Ashkenazi犹太人CMT1A家系中,每个受累个体在17p11.2内部存在大的DNA片段的重复(重复是指染色体DNA中某一区段或基因出现双倍拷贝的现象),重复大小约为1.5百万碱基(megabase,Mb),其两翼有长约17~29千碱基(kilobaseKb)的REP重复序列。另一组研究人员[5]通过对12个欧洲CMT1A家系的调查,也同时证明了该重复的存在。在以后的两年中,研究者们发现不同种族人群的CMT1A家系、不相关的CMT1患者及散发的CMT1患者均有这种重复。Wise等[6]用3种不同的方法检测63个不相关的CMT1患者,发现68%的患者有重复;而Hoogendijk等[13]发现10例散发患者中,9例有重复。最近,欧洲进行了一次联合性调查,发现819例不相关CMT1患者中70.7%具有重复[10]。
, 百拇医药
2 PMP22基因是CMT1A的致病基因
DNA重复如何影响CMT1A基因表达而产生CMT1A表型的呢?研究者们曾设想了4种可能性:(1)重复区内1个或多个基因过度表达或称基因的剂量效应。(2)重复连接部位基因中断。(3)1个重复基因的显性突变。(4)在改变基因调节的重复区内基因座位的改变,即位置效应。随后通过以下事实证实了基因剂量效应是CMT1A表型的关键:(1)一些个体具有细胞遗传学上可见的17号染色体整个短臂或近端区域的明显重复。这些个体除了具有CMT1的电生理和临床表型外,还表现为17p三体综合征的特征。重复区域包含CMT1A的DNA重复序列,也包括其近端和远端的序列。(2)CMT1A患者在17p11.2内存在3个拷贝数,而正常人只有两个拷贝。(3)Lupski等[4]发现1例表型严重的纯合子患者,在17p11.2内存在4个拷贝数,其父母均CMT1A杂合子。
在证实CMT1A表型中起重要作用的候选基因中,研究患有震颤病的鼠有很大帮助。等位基因显性震颤鼠(Tr)和半显性震颤鼠(Tr-J)突变的表型与CMT1A个体突变表型很相似,人和鼠都有外周神经的髓磷质缺陷,包围外周神经的“葱球”结构和神经冲动传导障碍。1992年,研究者报道了Tr及Tr-J鼠突变体的PMP22基因的2个点突变,位于鼠基因组11号染色体上,与人17号染色体具有同源性,以后很快证实PMP22基因包含在CMT1A重复区中。
, 百拇医药
PMP22的可能致病作用是通过研究一个荷兰CMT1A家系连锁分析证明[7],致病基因位于17p,但未检测到重复,而来自该家系的患者具有与Tr-J鼠相同的PMP22突变。Roa等[8]对32个不相关的无重复的CMT1患者进行PMP22编码区突变检测,发现1例10岁男孩PMP22基因在公认的跨膜区发生突变,分析其家庭成员,发现该突变是自发的。具有突变或重复的患者临床表现相似。因此,得出结
论:PMP22基因是CMT1A的致病基因,点突变或重复均能导致CMT1表型。
PMP22基因编码膜相关髓鞘蛋白,其分子量为21824U(22kDa)。PMP22基因产物包括4个公认的跨膜区域,位于周围神经髓鞘致密部分。Yoshikawa等[9]分析5个家系CMT1A患者活检腓肠神经的PMP22mRNA表达,发现5例患者的PMP22mRNA表达明显高于其它脱髓鞘神经病,为PMP22基因剂量效应提供了直接证据。
, 百拇医药
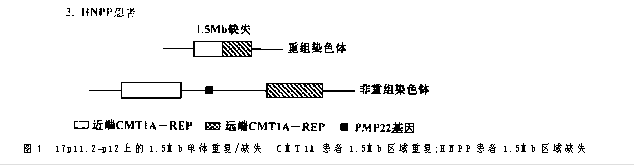
遗传性压力敏感性周围神经病(hereditary neuro pathy with liability to pressure palsies,HNPP)为另一常染色体显性遗传病,以反复发作的单神经或多神经麻痹为特征,常由轻微的外伤或挤压而诱发,腕管综合征和其它嵌压性神经病是HNPP常见的临床表现。电生理检查可见轻微的传导速度下降,亦可见传导阻滞。神经活检可见到节段性脱髓和髓鞘再生,在周围神经上形成“吞肠样”结构。HNPP的基因定位亦在17p11.2-p12,为1.5Mb区域缺失。CMT1A患者所有DNA重复遗传标记在HNPP患者中均缺失,而且缺失的界限与CMT1A重复连接界限相一致,说明该区域恰是CMT1A重复区域。HNPP亦可由PMP22阅读框架突变所引起。
研究人员推测HNPP中丢失的染色体和CMT1A重复的染色体是不平衡交换的相互产物。这种推测很快被证实:即分别观察到CMT1A、HNPP患者远端CMT1A-REP部分获得或丧失。因此建立起不平衡交换是重排的分子生物学基础(图1)。
, 百拇医药
3 重组热点的研究
Lopes等[10]为定位CMT1A-REPs内重组断点构建了近端和远端CMT1A-REP克隆EcoRⅠ片段,发现17个被检测的限制性位点中仅3个只存在于近端CMT1A-REP,而不存在于远端CMT1A-REP中,说明二者具有高度同源性。Kiyo-sawa等[14]通过对CMT1A-REPs内4Kb区域直接测序,证明二者的同源性为98%。因此,远端的CMT1A-REP与近端的CMT1A-REP可能发生错排,导致减数分裂期17号同源染色体发生不等臂交换。Lopes等分析了76例CMT1A和38例HNPP患者,将交换断点的热点定位于3.2Kb的区域,可解释75%的重排。用EcoRⅠ/SacⅠ消化后进行检测,CMT1A和HNPP患者分别存在3.2Kb和7.8Kb连接片段。而Reiter等[11]进一步将重组热点范围缩小,75%的CMT1A重复患者具有1.7KbEcoRⅠ/NsiⅠ连接片段,84%的HNPP缺失患者具有7.8KbEcoRⅠ/EcoRⅠ连接片段。Timmerman等[12]通过对不同种族不同人群研究得出不平衡交换的热点直接与CMT1A和HNPP患者发病机理相关的结论。而无重复的CMT1A患者和无缺失的HNPP患者均检测到PMP22基因突变,证明PMP22基因参与两种疾病的发病机理。
, 百拇医药
4 临床应用
上述的分子遗传学研究已应用于临床。17p重复和缺失的检测都很有意义,尤其是对无家族史的患者。基因重复的存在可鉴别CMT1A和其它脱髓鞘神经病,尤其是慢性炎症性脱髓鞘性多发性神经病;对已知有17p11.2重复的家系,可应用DNA诊断进行症状前和产前筛查。对儿童该项检查的重要意义在于鉴别受累儿童以便对他们经常检查,早期发现骨骼畸型,并通过适当的矫型治疗而避免。检测HNPP患者17p缺失比神经传导速度检查更敏感,因为并非所有患者都有临床或电生理改变;且比神经活检的损伤要小得多。检测患者家属同样有意义,有缺失者应特别注意保护周围神经。HNPP的早期诊断可使患者及时采取预防措施以避免某些部位神经受压或外伤,如肘部、腕部及腿的腓骨颈部等。
DNA分析用于产前诊断时只能检测或排除17p重复。而不能预示病情的严重程度,约20%患者到成年已明显残疾,但也有20%患者无症状,所以胎儿如有17p重复,夫妇双方即要求终止妊娠时才做产前检查。
当前诊断CMT1A重复/HNPP缺失的方法已有几种,包括限制性内切酶片段长度多态性;PCR扩增重复区域内多态性标记物;限制性酶切片段定量分析;脉冲场凝胶电泳检测特异性连接片段;有丝分裂中期染色体荧光原位杂交分析。PCR方法虽易于在一般实验室推广,但受杂和率的限制,且实验结果不稳定,需多次重复。其它方法亦受各种因素的限制,很难在临床推广,因此有必要建立一种快速、简便且诊断率高的方法应用于临床,这尚有待研究者进一步探索。
作者单位:100853北京,解放军总医院肌病室
参考文献(略), 百拇医药
关键词:进行性腓骨肌萎缩1A型 Charcot-Marie-Tooth病 遗传性运动感觉神经病
进行性腓骨肌萎缩症(Charcot-Marie-Tooth病,CMT),亦称为遗传性运动感觉神经病(hereditary motor and sen-sory neuropathy,HMSN),是神经系统常见的遗传性疾病之一,国外文献报道其发病率在1/2500~1/10000之间[1,2]。其中CMT1是最常见的亚型,其临床特点为:对称性肢体远端肌肉萎缩,慢性进行性力弱及弓形足畸形;电生理特征为运动感觉神经传导速度下降;组织化学可见髓神经纤维的数量减少,节段性脱髓鞘和髓鞘再生,形成典型的“洋葱球”结构。连锁分析证明CMT1是遗传异质性疾病,至少有4个位点。常染色体显性遗传的CMT1A、CMT1B和X连锁的显性CMTX。他们分别定位于17p11.2、1q21.3-23和Xq13,此外,CMT1C尚未定位。CMT1A是CMT1中最常见的类型,也是近年来研究的热点,我们就其分子生物学研究进展及临床应用情况综述如下。
, http://www.100md.com
1 基因序列重复片段的发现
1989年,Raeymaekers等[3]首次通过连锁分析将CMT1A的基因定位于17p11.2。随后于1991年Lupski等[4]发现在6个大French-AcadianCMT1A家系和1个Ashkenazi犹太人CMT1A家系中,每个受累个体在17p11.2内部存在大的DNA片段的重复(重复是指染色体DNA中某一区段或基因出现双倍拷贝的现象),重复大小约为1.5百万碱基(megabase,Mb),其两翼有长约17~29千碱基(kilobaseKb)的REP重复序列。另一组研究人员[5]通过对12个欧洲CMT1A家系的调查,也同时证明了该重复的存在。在以后的两年中,研究者们发现不同种族人群的CMT1A家系、不相关的CMT1患者及散发的CMT1患者均有这种重复。Wise等[6]用3种不同的方法检测63个不相关的CMT1患者,发现68%的患者有重复;而Hoogendijk等[13]发现10例散发患者中,9例有重复。最近,欧洲进行了一次联合性调查,发现819例不相关CMT1患者中70.7%具有重复[10]。
, 百拇医药
2 PMP22基因是CMT1A的致病基因
DNA重复如何影响CMT1A基因表达而产生CMT1A表型的呢?研究者们曾设想了4种可能性:(1)重复区内1个或多个基因过度表达或称基因的剂量效应。(2)重复连接部位基因中断。(3)1个重复基因的显性突变。(4)在改变基因调节的重复区内基因座位的改变,即位置效应。随后通过以下事实证实了基因剂量效应是CMT1A表型的关键:(1)一些个体具有细胞遗传学上可见的17号染色体整个短臂或近端区域的明显重复。这些个体除了具有CMT1的电生理和临床表型外,还表现为17p三体综合征的特征。重复区域包含CMT1A的DNA重复序列,也包括其近端和远端的序列。(2)CMT1A患者在17p11.2内存在3个拷贝数,而正常人只有两个拷贝。(3)Lupski等[4]发现1例表型严重的纯合子患者,在17p11.2内存在4个拷贝数,其父母均CMT1A杂合子。
在证实CMT1A表型中起重要作用的候选基因中,研究患有震颤病的鼠有很大帮助。等位基因显性震颤鼠(Tr)和半显性震颤鼠(Tr-J)突变的表型与CMT1A个体突变表型很相似,人和鼠都有外周神经的髓磷质缺陷,包围外周神经的“葱球”结构和神经冲动传导障碍。1992年,研究者报道了Tr及Tr-J鼠突变体的PMP22基因的2个点突变,位于鼠基因组11号染色体上,与人17号染色体具有同源性,以后很快证实PMP22基因包含在CMT1A重复区中。
, 百拇医药
PMP22的可能致病作用是通过研究一个荷兰CMT1A家系连锁分析证明[7],致病基因位于17p,但未检测到重复,而来自该家系的患者具有与Tr-J鼠相同的PMP22突变。Roa等[8]对32个不相关的无重复的CMT1患者进行PMP22编码区突变检测,发现1例10岁男孩PMP22基因在公认的跨膜区发生突变,分析其家庭成员,发现该突变是自发的。具有突变或重复的患者临床表现相似。因此,得出结
论:PMP22基因是CMT1A的致病基因,点突变或重复均能导致CMT1表型。
PMP22基因编码膜相关髓鞘蛋白,其分子量为21824U(22kDa)。PMP22基因产物包括4个公认的跨膜区域,位于周围神经髓鞘致密部分。Yoshikawa等[9]分析5个家系CMT1A患者活检腓肠神经的PMP22mRNA表达,发现5例患者的PMP22mRNA表达明显高于其它脱髓鞘神经病,为PMP22基因剂量效应提供了直接证据。
, 百拇医药
遗传性压力敏感性周围神经病(hereditary neuro pathy with liability to pressure palsies,HNPP)为另一常染色体显性遗传病,以反复发作的单神经或多神经麻痹为特征,常由轻微的外伤或挤压而诱发,腕管综合征和其它嵌压性神经病是HNPP常见的临床表现。电生理检查可见轻微的传导速度下降,亦可见传导阻滞。神经活检可见到节段性脱髓和髓鞘再生,在周围神经上形成“吞肠样”结构。HNPP的基因定位亦在17p11.2-p12,为1.5Mb区域缺失。CMT1A患者所有DNA重复遗传标记在HNPP患者中均缺失,而且缺失的界限与CMT1A重复连接界限相一致,说明该区域恰是CMT1A重复区域。HNPP亦可由PMP22阅读框架突变所引起。
研究人员推测HNPP中丢失的染色体和CMT1A重复的染色体是不平衡交换的相互产物。这种推测很快被证实:即分别观察到CMT1A、HNPP患者远端CMT1A-REP部分获得或丧失。因此建立起不平衡交换是重排的分子生物学基础(图1)。
, 百拇医药
3 重组热点的研究
Lopes等[10]为定位CMT1A-REPs内重组断点构建了近端和远端CMT1A-REP克隆EcoRⅠ片段,发现17个被检测的限制性位点中仅3个只存在于近端CMT1A-REP,而不存在于远端CMT1A-REP中,说明二者具有高度同源性。Kiyo-sawa等[14]通过对CMT1A-REPs内4Kb区域直接测序,证明二者的同源性为98%。因此,远端的CMT1A-REP与近端的CMT1A-REP可能发生错排,导致减数分裂期17号同源染色体发生不等臂交换。Lopes等分析了76例CMT1A和38例HNPP患者,将交换断点的热点定位于3.2Kb的区域,可解释75%的重排。用EcoRⅠ/SacⅠ消化后进行检测,CMT1A和HNPP患者分别存在3.2Kb和7.8Kb连接片段。而Reiter等[11]进一步将重组热点范围缩小,75%的CMT1A重复患者具有1.7KbEcoRⅠ/NsiⅠ连接片段,84%的HNPP缺失患者具有7.8KbEcoRⅠ/EcoRⅠ连接片段。Timmerman等[12]通过对不同种族不同人群研究得出不平衡交换的热点直接与CMT1A和HNPP患者发病机理相关的结论。而无重复的CMT1A患者和无缺失的HNPP患者均检测到PMP22基因突变,证明PMP22基因参与两种疾病的发病机理。
, 百拇医药
4 临床应用
上述的分子遗传学研究已应用于临床。17p重复和缺失的检测都很有意义,尤其是对无家族史的患者。基因重复的存在可鉴别CMT1A和其它脱髓鞘神经病,尤其是慢性炎症性脱髓鞘性多发性神经病;对已知有17p11.2重复的家系,可应用DNA诊断进行症状前和产前筛查。对儿童该项检查的重要意义在于鉴别受累儿童以便对他们经常检查,早期发现骨骼畸型,并通过适当的矫型治疗而避免。检测HNPP患者17p缺失比神经传导速度检查更敏感,因为并非所有患者都有临床或电生理改变;且比神经活检的损伤要小得多。检测患者家属同样有意义,有缺失者应特别注意保护周围神经。HNPP的早期诊断可使患者及时采取预防措施以避免某些部位神经受压或外伤,如肘部、腕部及腿的腓骨颈部等。
DNA分析用于产前诊断时只能检测或排除17p重复。而不能预示病情的严重程度,约20%患者到成年已明显残疾,但也有20%患者无症状,所以胎儿如有17p重复,夫妇双方即要求终止妊娠时才做产前检查。
当前诊断CMT1A重复/HNPP缺失的方法已有几种,包括限制性内切酶片段长度多态性;PCR扩增重复区域内多态性标记物;限制性酶切片段定量分析;脉冲场凝胶电泳检测特异性连接片段;有丝分裂中期染色体荧光原位杂交分析。PCR方法虽易于在一般实验室推广,但受杂和率的限制,且实验结果不稳定,需多次重复。其它方法亦受各种因素的限制,很难在临床推广,因此有必要建立一种快速、简便且诊断率高的方法应用于临床,这尚有待研究者进一步探索。
作者单位:100853北京,解放军总医院肌病室
参考文献(略), 百拇医药