Alport综合征三个系13例临床分析
作者:黄基格 林治瓯
单位:浙江省平阳县人民医院内科(325400)
关键词:
温州医学院学报/990225 Alport综合征是一以基底膜改变为特征的家族性遗传性疾病,临床较少见。现将三个家系13例患者资料报道如下。
1 临床资料
三个家系13例患者中,男9例,女4例,年龄17~52岁,平均31.5岁。家族甲:6例,其中男4例,女2例。6例均有血尿,其中5例蛋白尿阳性,4例神经性耳聋,近视及球型晶体各1例。4例因尿毒症已死亡。先证者男,38岁,父母系近亲结婚,主诉腰痛十余年,体检示血压18.7/10.7kPa,球型晶体,高频对称性神经耳聋,颜面下肢无浮肿。辅助检查:尿蛋白( )、尿红细胞(+),血肌酐763μmol/L,血尿素氮21.7mmol/L。家族乙:5例,其中男3例,女2例。5例均有血尿,3例伴蛋白尿,神经性耳聋2例,球型晶体1例,3例因尿毒症已死亡。先证者男,34岁,父母系近亲结婚,主诉反复肉眼血尿15年余,体检示血压22/12kPa,高频对称性神经性耳聋,颜面下肢稍浮肿。辅助检查:尿蛋白(),尿红细胞(
)、尿红细胞(+),血肌酐763μmol/L,血尿素氮21.7mmol/L。家族乙:5例,其中男3例,女2例。5例均有血尿,3例伴蛋白尿,神经性耳聋2例,球型晶体1例,3例因尿毒症已死亡。先证者男,34岁,父母系近亲结婚,主诉反复肉眼血尿15年余,体检示血压22/12kPa,高频对称性神经性耳聋,颜面下肢稍浮肿。辅助检查:尿蛋白(),尿红细胞( ),血肌酐824μmol/L,血尿素氮23.2mmol/L。家族丙:2例,均为男,2例均有蛋白尿及血尿,1例神经性耳聋,1例近视。先证者24岁,体检发现血压20/12kPa,高频对称神经性耳聋,颜面下肢无浮肿。辅助检查:尿蛋白(),尿红细胞(),血肌酐744μmol/L,血尿素氮22.1mmol/L。肾穿刺活检病理报告示:LM――肾皮质组织两小条,共见40个肾小球,大部分呈肾小球球性硬化,系膜弥漫增生,肾小管基底膜增厚,间质可见泡沫细胞。符合Alport综合征。家系谱见附图。
),血肌酐824μmol/L,血尿素氮23.2mmol/L。家族丙:2例,均为男,2例均有蛋白尿及血尿,1例神经性耳聋,1例近视。先证者24岁,体检发现血压20/12kPa,高频对称神经性耳聋,颜面下肢无浮肿。辅助检查:尿蛋白(),尿红细胞(),血肌酐744μmol/L,血尿素氮22.1mmol/L。肾穿刺活检病理报告示:LM――肾皮质组织两小条,共见40个肾小球,大部分呈肾小球球性硬化,系膜弥漫增生,肾小管基底膜增厚,间质可见泡沫细胞。符合Alport综合征。家系谱见附图。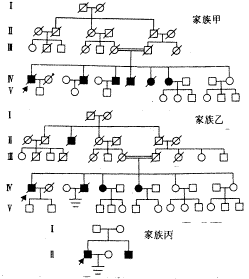
, 百拇医药
附图 三个家系图谱
□ ○ 健康者(□=男,○=女) ■ ● 患本病死亡者
■ ● 患病者 ■先证者 □ ○ 其它原因死亡者
2 讨论
Alport综合征有三种遗传方式,即X性连锁显性遗传,常染色体显性遗传,常染色体隐性遗传。它的特点是:①患者双亲表现型正常,但均为致病基因的肯定携带者。②有的系谱中只有先证者,系谱中看不到连续遗传现象,常为散发。③患者大部分出现在同胞之间,子女往往正常。④近亲婚配的后代中发病率显著增多[1,2]。据此,本组资料中甲乙两家系为常染色体隐性遗传。家族丙因患者故意隐瞒家族史,未能确定其遗传方式。
Alport综合征的诊断应包括临床―病理及基因两个水平的诊断[2]。在临床―病理水平诊断上,标准并未统一。Flinter等建议采用以下四条标准:①阳性家族史;②肾病变、血尿及进行性肾功能损害,并有肾小球基底膜的典型改变;③耳病变、高频对称性耳聋;④眼病变、球型或锥型晶体。只要具备以上的三条就能诊断。本组资料患者均符合此诊断标准,但在初诊时均误诊为急性或慢性肾炎。误诊原因如下:①本病肾脏表现无特异性,蛋白尿、血尿为肾炎共同表现;②本病为少见病,基层医院医师对其认识不足,肾外表现未引起医患双方重视;③未普及肾穿刺活检及免疫荧光检查;④有些患者故意隐瞒家庭史。由于Alport综合征为遗传性疾病,故无特效治疗方法。本组患者在劳累或上感后血尿加重肾功能恶化,故应注意避免。家族丙2例患者于三年前行肾移植手术治疗,且目前存活良好,不失为目前治疗Alport综合征的一种较好方法。
3 参考文献
[1] 李璞主编.医用生物学.第2版.北京:人民卫生出版社,1989.142
[2] 谌贻璞.Alport综合征及其研究现状.中华肾脏病杂志,1995,11(2)∶110
(收稿:1998-02-23,修回:1998-07-13), 百拇医药
单位:浙江省平阳县人民医院内科(325400)
关键词:
温州医学院学报/990225 Alport综合征是一以基底膜改变为特征的家族性遗传性疾病,临床较少见。现将三个家系13例患者资料报道如下。
1 临床资料
三个家系13例患者中,男9例,女4例,年龄17~52岁,平均31.5岁。家族甲:6例,其中男4例,女2例。6例均有血尿,其中5例蛋白尿阳性,4例神经性耳聋,近视及球型晶体各1例。4例因尿毒症已死亡。先证者男,38岁,父母系近亲结婚,主诉腰痛十余年,体检示血压18.7/10.7kPa,球型晶体,高频对称性神经耳聋,颜面下肢无浮肿。辅助检查:尿蛋白(
)、尿红细胞(+),血肌酐763μmol/L,血尿素氮21.7mmol/L。家族乙:5例,其中男3例,女2例。5例均有血尿,3例伴蛋白尿,神经性耳聋2例,球型晶体1例,3例因尿毒症已死亡。先证者男,34岁,父母系近亲结婚,主诉反复肉眼血尿15年余,体检示血压22/12kPa,高频对称性神经性耳聋,颜面下肢稍浮肿。辅助检查:尿蛋白(),尿红细胞(),血肌酐824μmol/L,血尿素氮23.2mmol/L。家族丙:2例,均为男,2例均有蛋白尿及血尿,1例神经性耳聋,1例近视。先证者24岁,体检发现血压20/12kPa,高频对称神经性耳聋,颜面下肢无浮肿。辅助检查:尿蛋白(),尿红细胞(),血肌酐744μmol/L,血尿素氮22.1mmol/L。肾穿刺活检病理报告示:LM――肾皮质组织两小条,共见40个肾小球,大部分呈肾小球球性硬化,系膜弥漫增生,肾小管基底膜增厚,间质可见泡沫细胞。符合Alport综合征。家系谱见附图。, 百拇医药
附图 三个家系图谱
□ ○ 健康者(□=男,○=女) ■ ● 患本病死亡者
■ ● 患病者 ■先证者 □ ○ 其它原因死亡者
2 讨论
Alport综合征有三种遗传方式,即X性连锁显性遗传,常染色体显性遗传,常染色体隐性遗传。它的特点是:①患者双亲表现型正常,但均为致病基因的肯定携带者。②有的系谱中只有先证者,系谱中看不到连续遗传现象,常为散发。③患者大部分出现在同胞之间,子女往往正常。④近亲婚配的后代中发病率显著增多[1,2]。据此,本组资料中甲乙两家系为常染色体隐性遗传。家族丙因患者故意隐瞒家族史,未能确定其遗传方式。
Alport综合征的诊断应包括临床―病理及基因两个水平的诊断[2]。在临床―病理水平诊断上,标准并未统一。Flinter等建议采用以下四条标准:①阳性家族史;②肾病变、血尿及进行性肾功能损害,并有肾小球基底膜的典型改变;③耳病变、高频对称性耳聋;④眼病变、球型或锥型晶体。只要具备以上的三条就能诊断。本组资料患者均符合此诊断标准,但在初诊时均误诊为急性或慢性肾炎。误诊原因如下:①本病肾脏表现无特异性,蛋白尿、血尿为肾炎共同表现;②本病为少见病,基层医院医师对其认识不足,肾外表现未引起医患双方重视;③未普及肾穿刺活检及免疫荧光检查;④有些患者故意隐瞒家庭史。由于Alport综合征为遗传性疾病,故无特效治疗方法。本组患者在劳累或上感后血尿加重肾功能恶化,故应注意避免。家族丙2例患者于三年前行肾移植手术治疗,且目前存活良好,不失为目前治疗Alport综合征的一种较好方法。
3 参考文献
[1] 李璞主编.医用生物学.第2版.北京:人民卫生出版社,1989.142
[2] 谌贻璞.Alport综合征及其研究现状.中华肾脏病杂志,1995,11(2)∶110
(收稿:1998-02-23,修回:1998-07-13), 百拇医药