Hallervorden-Spatz病一例
作者:常杏芝 张月华 包新华
单位:常杏芝(北京医科大学第一医院儿科 100034);张月华(北京医科大学第一医院儿科 100034);包新华(北京医科大学第一医院儿科 100034)
关键词:
中华儿科杂志000127 患儿男,12岁。因进行性走路不稳5年半,抽搐1次入院。5年半前患儿出现抽搐1次,表现为全身强直发作,持续约10 min。之后家长发现患儿逐渐走路不稳,时有跌倒,表现为突然的身体向后倾倒,能立即自行站起。4年半前逐渐出现双上肢关节交替屈曲、伸展、扭转,以左上肢腕关节为著,睡眠中消失。4年前渐出现言语不清。半年前,走路时频繁摔倒,不能独走,扶走时双下肢不灵活,四肢不自主运动频繁,言语不清明显加重。发病后反应较前迟钝,智力倒退不明显。体检:神志清,面部表情呆板,构音不清,步态不稳,双上肢不自主舞动,躯干轻度后仰,时有右下肢不自主踢动。心、肺、腹部未见异常。四肢肌力正常,肌张力增高,以左上肢为著,肢体无震颤,指鼻动作准确,不能走直线,Romberg征(-),双膝腱反射、跟腱反射活跃,踝阵挛(+),双侧Barbinski征(+)。角膜K-F环阴性。辅助检查:肝、肾功能正常,血气分析正常,血乳酸、丙酮酸最小运动量试验正常,血铜蓝蛋白正常,EEG正常。智测:IQ 52分。头颅MRI示双侧苍白球区T1W呈等信号,T2W呈中央对称性高信号,周边呈对称性低信号(虎眼征)(图1)。诊断为Hallervorden-Spatz病,给予小剂量氟哌啶醇与安坦治疗,2周后不自主运动减少,余症状无明显改善,出院观察。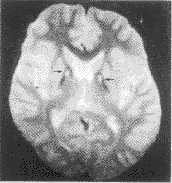
, http://www.100md.com
图1 T2W像 示双侧苍白球区呈中央对称性高信号,周边呈对称性低信号(↑)
讨论:Hallervorden-Spatz病,又名苍白球黑质变性或婴儿晚期神经轴索营养不良,为罕见的遗传性铁代谢障碍性疾病,其遗传方式为常染色体隐性遗传,目前尚无确切的基因定位。该病突出的临床特征为锥体外系功能障碍,表现为肌张力不全,肌强直,舞蹈手足徐动症和构音障碍。多于20岁以前发病,半数以上的患儿于2~10岁出现症状,临床症状进行性加重。可有皮质脊髓束受累,出现巴氏征阳性,深腱反射活跃。20%~25%的病人可有色素性视网膜炎和惊厥,及进行性智力损害。本病预后不良,病程一般5~10年,多于成年早期死亡。本病常规检查无异常,同位素检查发现基底节对59Fe摄取增加,皮肤成纤维细胞培养可见转铁蛋白积聚,头颅MRI T2W示苍白球区有“虎眼征”是其特征性表现。神经病理检查可见苍白球、黑质区铁质沉着变性及蜡样质脂褐素和神经黑素沉积,苍白球区神经元减少、髓鞘变薄。脑皮质及苍白球黑质系统见广泛播散的局灶性神经轴球形肿胀,与婴儿期神经轴营养不良病理改变相似,推测二者之间可能有等位基因关系。
本病的诊断主要依据典型临床表现及特征性的头颅MRI改变。鉴别诊断需首先除外静止性病程的锥体外系疾病,如脑性瘫痪、一氧化碳或霉变甘蔗中毒、以及药物引起的锥体外系症状。进行性病程者需除外肝豆状核变性,Huntington舞蹈病,变形性肌张力不全及少年型蜡样质脂褐质沉积症。本病目前主要是对症治疗。
收稿日期:1999-02-14, 百拇医药
单位:常杏芝(北京医科大学第一医院儿科 100034);张月华(北京医科大学第一医院儿科 100034);包新华(北京医科大学第一医院儿科 100034)
关键词:
中华儿科杂志000127 患儿男,12岁。因进行性走路不稳5年半,抽搐1次入院。5年半前患儿出现抽搐1次,表现为全身强直发作,持续约10 min。之后家长发现患儿逐渐走路不稳,时有跌倒,表现为突然的身体向后倾倒,能立即自行站起。4年半前逐渐出现双上肢关节交替屈曲、伸展、扭转,以左上肢腕关节为著,睡眠中消失。4年前渐出现言语不清。半年前,走路时频繁摔倒,不能独走,扶走时双下肢不灵活,四肢不自主运动频繁,言语不清明显加重。发病后反应较前迟钝,智力倒退不明显。体检:神志清,面部表情呆板,构音不清,步态不稳,双上肢不自主舞动,躯干轻度后仰,时有右下肢不自主踢动。心、肺、腹部未见异常。四肢肌力正常,肌张力增高,以左上肢为著,肢体无震颤,指鼻动作准确,不能走直线,Romberg征(-),双膝腱反射、跟腱反射活跃,踝阵挛(+),双侧Barbinski征(+)。角膜K-F环阴性。辅助检查:肝、肾功能正常,血气分析正常,血乳酸、丙酮酸最小运动量试验正常,血铜蓝蛋白正常,EEG正常。智测:IQ 52分。头颅MRI示双侧苍白球区T1W呈等信号,T2W呈中央对称性高信号,周边呈对称性低信号(虎眼征)(图1)。诊断为Hallervorden-Spatz病,给予小剂量氟哌啶醇与安坦治疗,2周后不自主运动减少,余症状无明显改善,出院观察。
, http://www.100md.com
图1 T2W像 示双侧苍白球区呈中央对称性高信号,周边呈对称性低信号(↑)
讨论:Hallervorden-Spatz病,又名苍白球黑质变性或婴儿晚期神经轴索营养不良,为罕见的遗传性铁代谢障碍性疾病,其遗传方式为常染色体隐性遗传,目前尚无确切的基因定位。该病突出的临床特征为锥体外系功能障碍,表现为肌张力不全,肌强直,舞蹈手足徐动症和构音障碍。多于20岁以前发病,半数以上的患儿于2~10岁出现症状,临床症状进行性加重。可有皮质脊髓束受累,出现巴氏征阳性,深腱反射活跃。20%~25%的病人可有色素性视网膜炎和惊厥,及进行性智力损害。本病预后不良,病程一般5~10年,多于成年早期死亡。本病常规检查无异常,同位素检查发现基底节对59Fe摄取增加,皮肤成纤维细胞培养可见转铁蛋白积聚,头颅MRI T2W示苍白球区有“虎眼征”是其特征性表现。神经病理检查可见苍白球、黑质区铁质沉着变性及蜡样质脂褐素和神经黑素沉积,苍白球区神经元减少、髓鞘变薄。脑皮质及苍白球黑质系统见广泛播散的局灶性神经轴球形肿胀,与婴儿期神经轴营养不良病理改变相似,推测二者之间可能有等位基因关系。
本病的诊断主要依据典型临床表现及特征性的头颅MRI改变。鉴别诊断需首先除外静止性病程的锥体外系疾病,如脑性瘫痪、一氧化碳或霉变甘蔗中毒、以及药物引起的锥体外系症状。进行性病程者需除外肝豆状核变性,Huntington舞蹈病,变形性肌张力不全及少年型蜡样质脂褐质沉积症。本病目前主要是对症治疗。
收稿日期:1999-02-14, 百拇医药