(1ЁфR,2S,2ЁфS)-2-(2-МзЛљ-2-фхМзЛљ-3-бЧМзЛљ)ЛЗЮьЛљБћЫсЕФКЯГЩ
зїепЃКЙмУЕ ЮтгТ*
ЕЅЮЛЃКЛЊЮївНПЦДѓбЇИНвЛдКЃЌвЉбЇдК* ГЩЖМ 610041
ЙиМќДЪЃК(+)-ФкЯђ-3-фхнЈЭЊ-2ЃЛЙтбЇЛюадЃЛ(1ЁфRЃЌ2SЃЌ2ЁфS)-2-(2-МзЛљ-2-фхМзЛљ-3-бЧМзЛљ)ЛЗЮьЛљБћЫсЃЛКЯГЩ
ЛЊЮївЉбЇдгжО990411 ЬсвЊ вдЩЬвЕвзЕУЕФ(+)-ФкЯђ-3-фхнЈЭЊ-2ЮЊЪжаддДКЯГЩСЫЙтбЇЛюадЕФ(1ЁфRЃЌ2SЃЌ2ЁфS)-2-
(2-фхМзЛљ-2-МзЛљ-3-бЧМзЛљ)ЛЗЮьЛљБћЫсЁЃ
THE SYNTHESIS OF (1ЁфRЃЌ2SЃЌ2ЁфS)-2-(2-BROMOMETHYL-2-
METHYL-3-METHYLENE)CYCLOPENTYLPROPIONIC ACID
, АйФДвНвЉ
Guan Mei Wu Yong*
(The First Affiliated Hospital, School of Pharmacy*,West China University of Medical Scineces Chengdu 610041)
ABSTRACT A method for synthesis of optically active (1ЁфR, 2S, 2ЁфS)-2-(2
-bromomethyl-2-methyl-3-methylene) cyclopentylpropionic acid starting from commercial available(+)-3-Endo-Bromocamphor is described.
Key Words (+)-3-Endo-bromocamphor Optical activity (1ЁфR,2S,2ЁфS)-2-(2
, АйФДвНвЉ
-Bromomethyl-2-methyl-3-methylene)cyclopentylpropionic acid Synthesis
вдЩЬвЕвзЕУЕФ(+)-ФкЯђ-3-фхнЈЭЊ-2(1)ЮЊЪжаддДРДКЯГЩФГаЉЬьШЛЛЏКЯЮяКЭЪжадКЯГЩзгЕФбаОПвбЪмЕНШЫУЧЕФжиЪг[1ЃЌ2]ЁЃдкбаОПVitamin D3РрЫЦЮяЕФШЋКЯГЩЪБЃЌвВМЦЛЎВЩгУ(+)-ФкЯђ-3-фхнЈЭЊ-2(1)ЮЊЪжаддДРДКЯГЩЙиМќжаМфЬх(1ЁфRЃЌ2SЃЌ2ЁфS)-2-(2-МзЛљ2-фхМзЛљ-3-бЧМзЛљ)ЛЗЮьЛљБћЫсЃЌВЂЩшМЦСЫШчScheme 1ЫљЪОЕФКЯГЩТЗЯпЁЃ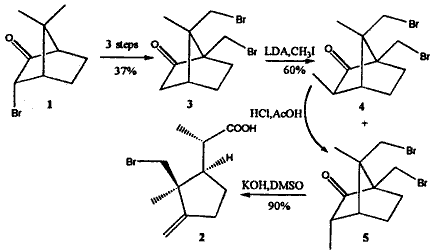
MoneyЕШШЫ[2ЃЌ3]дјБЈЕРЙ§(1)зЊЛЏГЩ(+)-ЭтЯђ-3-МзЛљ-9ЃЌ10-ЖўфхнЈЭЊ(4)КЭЫќЕФВюЯђвьЙЙЬх(5)ЕФЗНЗЈЃЌЕЋЙиМќвЛВН(+)-9ЃЌ10-ЖўфхнЈЭЊ(3)зЊЛЏГЩ(4)КЭ(5)ЕФЗДгІЪеТЪНі34%ЁЃЪЕбщЪзЯШАДMoneyЕШШЫ[2]ЕФЗНЗЈЃЌвд(1)ЮЊдСЯЃЌО3ВНКЯГЩЛЏКЯЮязмЪеТЪЮЊ37%ЃЌЫцКѓЯъЯИЕиЬНЬжСЫ(3)зЊЛЏГЩ(4)КЭ(5)ЕФЬѕМўЃЌВЂЗЂЯжЃЌЕБ(3)гыLDAЁЂHMPAКЭЕтМзЭщЕФФІЖћБШЮЊ1ЁУ1.2ЁУ1.3ЁУ5ЃЌдк4ЁцЗДгІ3 hКѓЃЌЦфЪеТЪПЩДя60%ЃЌ500 MHz NMRЯдЪО(4)КЭ(5)ЕФБШР§ЮЊ1.8ЁУ1ЁЃгЩгк(5)ЪЧШШСІбЇИќЮШЖЈЕФвьЙЙЬх[3]ЃЌвђДЫЃЌНЋБШР§ЮЊ1.8ЁУ1ЕФ(4)КЭ(5)ЕФЛьКЯЮядкбЮЫсКЭДзЫсЛьКЯвКжаЛиСї16 hКѓЃЌЕУЕНБШР§ЮЊ1ЁУ6ЕФ(4)КЭ(5)ЪеТЪ90%ЃЌОжљВуЮіКЭЖрДЮжиНсОЇКѓЃЌЕУЙтбЇДПЕФ(5)ЃЌСНВНЪеТЪПЩДя42%ЁЃдйНЋЙтбЇДПЕФ(5)дкМюДпЛЏЯТПЊЛЗЃЌМДЕУФПБъЮя(2)ЃЌЦфНсЙЙОIRЁЂ1H NMRКЭдЊЫиЗжЮіШЗжЄЁЃ
, АйФДвНвЉ
1 ЪЕбщВПЗж
1.1 вЧЦїКЭЪдМС
ЫљгУЪдМСКЭШмМСЮЊЗжЮіДПЃЌгУЧААДвбжЊЗНЗЈДПЛЏКЭИЩдя[4]ЁЃШлЕугУX-4аЭЯдЮЂШлЕувЧВтЖЈЃЌЮДаЃе§ЃЛа§ЙтЖШгУPerkin-Elmer 241аЭвЧВтЖЈЃЛIRгУPerkin-Elmer 983аЭвЧВтЖЈЃЌKBrбЙЦЌЗЈЃЛ1H NMRгУAM-500аЭвЧВтЖЈЃЌCDCl3ЮЊШмМСЃЌTMSЮЊФкБъЃЛдЊЫиЗжЮігУCarl-Erba 1106аЭвЧВтЖЈЁЃ
1.2 КЯГЩЗНЗЈ
1.2.1 (+)-9ЃЌ10-ЖўфхнЈЭЊ-2(3)ЕФКЯГЩ ВЮееЮФЯзЗНЗЈ[2]вд(+)-ФкЯђ-3-фхнЈЭЊ-2ЮЊдСЯЃЌО3ВНЕУзмЪеТЪЮЊ37%ЕФАзЩЋЙЬЬх(3)ЃЌmp (98ЁЋ99)ЁцЃЌ[ІС]20D68.5Ёу(C 1.4ЃЌCHCl3)ЃЌЮФЯз[2]mp (97ЁЋ98)ЁцЃЌ[ІС]22D67Ёу(C 1.1ЃЌCHCl3)ЁЃ
, АйФДвНвЉ
1.2.2 (+)-ФкЯђ-3-МзЛљ-9ЃЌ10-ЖўфхнЈЭЊ-2(5)ЕФКЯГЩ ЛьКЯЖўвьБћАЗ7 ml(52.3 mmol)КЭЮоЫЎЫФЧтпЛрЋ100 mlЃЌЭЈыВЦјЯТЃЌгк0ЁцЕЮМге§ЖЁЛљяЎЕФе§МКЭщвК19 ml(2.5 mol/L)ЃЌНСАш40 minКѓЃЌЕЮМг(3)12.3 g(39.5 mmol)ЕФЮоЫЎЫФЧтпЛрЋвК50 mlЃЌHMPA9 mlКЭЕтМзЭщ28 g(198 mmol)ЃЌКуЮТгк4ЁцНСАш3 hКѓЃЌГхШы100 mlБљЫЎжаЃЌгУввУб500 mlЗжСНДЮЬсШЁЃЌКЯВЂЬсШЁвКЃЌЮоЫЎСђЫсФЦИЩдяЃЌЙшНКПьЫйжљВуЮіЃЌЕУБШР§ЮЊ1ЁУ1.8ЕФАзЩЋЙЬЬхЛьКЯЮя(5)КЭ(4)7.6 gЃЌЪеТЪ60%ЃЌВЂЛжИД(3)4 gЁЃВЮееЮФЯзЗНЗЈ[3]ЃЌгУХЈбЮЫсКЭДзЫсДІРэ(5)КЭ(4)ЕФЛьКЯЮяЃЌОКѓДІРэКЭДПЛЏЕУ5.2 gЙтбЇДПЕФ(5)ЃЌСНВНЪеТЪ42%ЁЃmp 63ЁЋ64ЁцЃЌ[ІС]20D+47Ёу(C 0.25ЃЌCH2Cl2)ЃЌЮФЯзmp 62.5ЁЋ
63ЁцЃЌ[ІС]22D+46.7Ёу(C 0.30ЃЌCH2Cl2)ЁЃIRcm-1ЃК2981ЃЌ1731ЁЃ1H NMR ІФЃК1.11(3HЃЌd,J=
, АйФДвНвЉ
8Hz)ЃЌ1.16(3HЃЌs)ЃЌ2.43ЁЋ2.54(2H,m),4.44ЁЋ4.66(2HЃЌAB quartet,J=12Hz)ЃЌ4.61ЁЋ4.74
(2HЃЌAB quartet,J=10Hz)ЁЃ
1.2.3 (1ЁфRЃЌ2SЃЌ2ЁфS)-2-(2-МзЛљ-2-фхМзЛљ-3-бЧМзЛљ)ЛЗЮьЛљБћЫс(2)ЕФКЯГЩ ЛьКЯ(5)1.56 g(4.8 mmol)ЁЂЧтбѕЛЏМи1.35 g(24 mmol)ЁЂЖўМзбЧэП51 mlКЭЫЎ10 mlЃЌЪвЮТНСАш1 hЃЌНЋДЫЗДгІвКГхШы20 mlБљЫЎжаЃЌввУб100 mlЯДЃЌЫЎВугУХЈбЮЫсЫсЛЏжСpH 2ЁЋ3ЃЌгУввУб200 mlЗжЫФДЮЬсШЁЃЌКЯВЂЬсШЁвКЃЌЮоЫЎСђЫсФЦИЩдяЃЌОМѕбЙХЈЫѕЕУ1.1g(2)ЃЌЪеТЪ90%ЁЃ[ІС]20D-81.2Ёу(C
1.1,CHCl3)ЁЃIRcm-1ЃК2550ЁЋ3300ЃЌ2964ЃЌ1707ЃЌ1653ЁЃ1H NMR 5.00(1H,s),4.71(1H,S),3.55ЁЋ
, АйФДвНвЉ
3.50(2H,d,J=10.5Hz),2.51(1H,dd,J=7.00,8.53Hz),2.40(2H,m),2.30(1H,m),1.80(1H,m),1.52
(1H,ddd,J=8.0,11.5,23.5Hz),1.31(3H,d,J=7.0Hz),1.10(3H,s)ЁЃдЊЫиЗжЮі(МЦЫужЕ/ЪЕВтжЕ%)ЃК
C 50.59/50.54ЃЌH 6.56/6.52ЁЃ
*ЭЈбЖСЊЯЕШЫ
ВЮПМЮФЯз
1 Hutchinson JH, Money TЃЌPiper SE. Ring cleavage of camphor derivatives:formation of chiral synthon for natural product synthesis.Can J.Chem, 1986, 64ЁУ854
, АйФДвНвЉ
2 Clase JAЃЌMoney T. An enantiospecific route to C, D ring synthon for steroid synthesis. Can J.Chem, 1992, 70ЁУ1537
3 Hutchinson JHЃЌ Money T. Stereoselectivity of C(3)-methylation of camphor and derivatives. Can J. Chem, 1984, 62ЁУ1899
4 Li SWЃЌ Fan RL. Handbook of practical chemistry. Shanghai:Shanghai sciences and technology press. 1981, 549, АйФДвНвЉ(ЙмУЕ ЮтгТ*)
ЕЅЮЛЃКЛЊЮївНПЦДѓбЇИНвЛдКЃЌвЉбЇдК* ГЩЖМ 610041
ЙиМќДЪЃК(+)-ФкЯђ-3-фхнЈЭЊ-2ЃЛЙтбЇЛюадЃЛ(1ЁфRЃЌ2SЃЌ2ЁфS)-2-(2-МзЛљ-2-фхМзЛљ-3-бЧМзЛљ)ЛЗЮьЛљБћЫсЃЛКЯГЩ
ЛЊЮївЉбЇдгжО990411 ЬсвЊ вдЩЬвЕвзЕУЕФ(+)-ФкЯђ-3-фхнЈЭЊ-2ЮЊЪжаддДКЯГЩСЫЙтбЇЛюадЕФ(1ЁфRЃЌ2SЃЌ2ЁфS)-2-
(2-фхМзЛљ-2-МзЛљ-3-бЧМзЛљ)ЛЗЮьЛљБћЫсЁЃ
THE SYNTHESIS OF (1ЁфRЃЌ2SЃЌ2ЁфS)-2-(2-BROMOMETHYL-2-
METHYL-3-METHYLENE)CYCLOPENTYLPROPIONIC ACID
, АйФДвНвЉ
Guan Mei Wu Yong*
(The First Affiliated Hospital, School of Pharmacy*,West China University of Medical Scineces Chengdu 610041)
ABSTRACT A method for synthesis of optically active (1ЁфR, 2S, 2ЁфS)-2-(2
-bromomethyl-2-methyl-3-methylene) cyclopentylpropionic acid starting from commercial available(+)-3-Endo-Bromocamphor is described.
Key Words (+)-3-Endo-bromocamphor Optical activity (1ЁфR,2S,2ЁфS)-2-(2
, АйФДвНвЉ
-Bromomethyl-2-methyl-3-methylene)cyclopentylpropionic acid Synthesis
вдЩЬвЕвзЕУЕФ(+)-ФкЯђ-3-фхнЈЭЊ-2(1)ЮЊЪжаддДРДКЯГЩФГаЉЬьШЛЛЏКЯЮяКЭЪжадКЯГЩзгЕФбаОПвбЪмЕНШЫУЧЕФжиЪг[1ЃЌ2]ЁЃдкбаОПVitamin D3РрЫЦЮяЕФШЋКЯГЩЪБЃЌвВМЦЛЎВЩгУ(+)-ФкЯђ-3-фхнЈЭЊ-2(1)ЮЊЪжаддДРДКЯГЩЙиМќжаМфЬх(1ЁфRЃЌ2SЃЌ2ЁфS)-2-(2-МзЛљ2-фхМзЛљ-3-бЧМзЛљ)ЛЗЮьЛљБћЫсЃЌВЂЩшМЦСЫШчScheme 1ЫљЪОЕФКЯГЩТЗЯпЁЃ
MoneyЕШШЫ[2ЃЌ3]дјБЈЕРЙ§(1)зЊЛЏГЩ(+)-ЭтЯђ-3-МзЛљ-9ЃЌ10-ЖўфхнЈЭЊ(4)КЭЫќЕФВюЯђвьЙЙЬх(5)ЕФЗНЗЈЃЌЕЋЙиМќвЛВН(+)-9ЃЌ10-ЖўфхнЈЭЊ(3)зЊЛЏГЩ(4)КЭ(5)ЕФЗДгІЪеТЪНі34%ЁЃЪЕбщЪзЯШАДMoneyЕШШЫ[2]ЕФЗНЗЈЃЌвд(1)ЮЊдСЯЃЌО3ВНКЯГЩЛЏКЯЮязмЪеТЪЮЊ37%ЃЌЫцКѓЯъЯИЕиЬНЬжСЫ(3)зЊЛЏГЩ(4)КЭ(5)ЕФЬѕМўЃЌВЂЗЂЯжЃЌЕБ(3)гыLDAЁЂHMPAКЭЕтМзЭщЕФФІЖћБШЮЊ1ЁУ1.2ЁУ1.3ЁУ5ЃЌдк4ЁцЗДгІ3 hКѓЃЌЦфЪеТЪПЩДя60%ЃЌ500 MHz NMRЯдЪО(4)КЭ(5)ЕФБШР§ЮЊ1.8ЁУ1ЁЃгЩгк(5)ЪЧШШСІбЇИќЮШЖЈЕФвьЙЙЬх[3]ЃЌвђДЫЃЌНЋБШР§ЮЊ1.8ЁУ1ЕФ(4)КЭ(5)ЕФЛьКЯЮядкбЮЫсКЭДзЫсЛьКЯвКжаЛиСї16 hКѓЃЌЕУЕНБШР§ЮЊ1ЁУ6ЕФ(4)КЭ(5)ЪеТЪ90%ЃЌОжљВуЮіКЭЖрДЮжиНсОЇКѓЃЌЕУЙтбЇДПЕФ(5)ЃЌСНВНЪеТЪПЩДя42%ЁЃдйНЋЙтбЇДПЕФ(5)дкМюДпЛЏЯТПЊЛЗЃЌМДЕУФПБъЮя(2)ЃЌЦфНсЙЙОIRЁЂ1H NMRКЭдЊЫиЗжЮіШЗжЄЁЃ
, АйФДвНвЉ
1 ЪЕбщВПЗж
1.1 вЧЦїКЭЪдМС
ЫљгУЪдМСКЭШмМСЮЊЗжЮіДПЃЌгУЧААДвбжЊЗНЗЈДПЛЏКЭИЩдя[4]ЁЃШлЕугУX-4аЭЯдЮЂШлЕувЧВтЖЈЃЌЮДаЃе§ЃЛа§ЙтЖШгУPerkin-Elmer 241аЭвЧВтЖЈЃЛIRгУPerkin-Elmer 983аЭвЧВтЖЈЃЌKBrбЙЦЌЗЈЃЛ1H NMRгУAM-500аЭвЧВтЖЈЃЌCDCl3ЮЊШмМСЃЌTMSЮЊФкБъЃЛдЊЫиЗжЮігУCarl-Erba 1106аЭвЧВтЖЈЁЃ
1.2 КЯГЩЗНЗЈ
1.2.1 (+)-9ЃЌ10-ЖўфхнЈЭЊ-2(3)ЕФКЯГЩ ВЮееЮФЯзЗНЗЈ[2]вд(+)-ФкЯђ-3-фхнЈЭЊ-2ЮЊдСЯЃЌО3ВНЕУзмЪеТЪЮЊ37%ЕФАзЩЋЙЬЬх(3)ЃЌmp (98ЁЋ99)ЁцЃЌ[ІС]20D68.5Ёу(C 1.4ЃЌCHCl3)ЃЌЮФЯз[2]mp (97ЁЋ98)ЁцЃЌ[ІС]22D67Ёу(C 1.1ЃЌCHCl3)ЁЃ
, АйФДвНвЉ
1.2.2 (+)-ФкЯђ-3-МзЛљ-9ЃЌ10-ЖўфхнЈЭЊ-2(5)ЕФКЯГЩ ЛьКЯЖўвьБћАЗ7 ml(52.3 mmol)КЭЮоЫЎЫФЧтпЛрЋ100 mlЃЌЭЈыВЦјЯТЃЌгк0ЁцЕЮМге§ЖЁЛљяЎЕФе§МКЭщвК19 ml(2.5 mol/L)ЃЌНСАш40 minКѓЃЌЕЮМг(3)12.3 g(39.5 mmol)ЕФЮоЫЎЫФЧтпЛрЋвК50 mlЃЌHMPA9 mlКЭЕтМзЭщ28 g(198 mmol)ЃЌКуЮТгк4ЁцНСАш3 hКѓЃЌГхШы100 mlБљЫЎжаЃЌгУввУб500 mlЗжСНДЮЬсШЁЃЌКЯВЂЬсШЁвКЃЌЮоЫЎСђЫсФЦИЩдяЃЌЙшНКПьЫйжљВуЮіЃЌЕУБШР§ЮЊ1ЁУ1.8ЕФАзЩЋЙЬЬхЛьКЯЮя(5)КЭ(4)7.6 gЃЌЪеТЪ60%ЃЌВЂЛжИД(3)4 gЁЃВЮееЮФЯзЗНЗЈ[3]ЃЌгУХЈбЮЫсКЭДзЫсДІРэ(5)КЭ(4)ЕФЛьКЯЮяЃЌОКѓДІРэКЭДПЛЏЕУ5.2 gЙтбЇДПЕФ(5)ЃЌСНВНЪеТЪ42%ЁЃmp 63ЁЋ64ЁцЃЌ[ІС]20D+47Ёу(C 0.25ЃЌCH2Cl2)ЃЌЮФЯзmp 62.5ЁЋ
63ЁцЃЌ[ІС]22D+46.7Ёу(C 0.30ЃЌCH2Cl2)ЁЃIRcm-1ЃК2981ЃЌ1731ЁЃ1H NMR ІФЃК1.11(3HЃЌd,J=
, АйФДвНвЉ
8Hz)ЃЌ1.16(3HЃЌs)ЃЌ2.43ЁЋ2.54(2H,m),4.44ЁЋ4.66(2HЃЌAB quartet,J=12Hz)ЃЌ4.61ЁЋ4.74
(2HЃЌAB quartet,J=10Hz)ЁЃ
1.2.3 (1ЁфRЃЌ2SЃЌ2ЁфS)-2-(2-МзЛљ-2-фхМзЛљ-3-бЧМзЛљ)ЛЗЮьЛљБћЫс(2)ЕФКЯГЩ ЛьКЯ(5)1.56 g(4.8 mmol)ЁЂЧтбѕЛЏМи1.35 g(24 mmol)ЁЂЖўМзбЧэП51 mlКЭЫЎ10 mlЃЌЪвЮТНСАш1 hЃЌНЋДЫЗДгІвКГхШы20 mlБљЫЎжаЃЌввУб100 mlЯДЃЌЫЎВугУХЈбЮЫсЫсЛЏжСpH 2ЁЋ3ЃЌгУввУб200 mlЗжЫФДЮЬсШЁЃЌКЯВЂЬсШЁвКЃЌЮоЫЎСђЫсФЦИЩдяЃЌОМѕбЙХЈЫѕЕУ1.1g(2)ЃЌЪеТЪ90%ЁЃ[ІС]20D-81.2Ёу(C
1.1,CHCl3)ЁЃIRcm-1ЃК2550ЁЋ3300ЃЌ2964ЃЌ1707ЃЌ1653ЁЃ1H NMR 5.00(1H,s),4.71(1H,S),3.55ЁЋ
, АйФДвНвЉ
3.50(2H,d,J=10.5Hz),2.51(1H,dd,J=7.00,8.53Hz),2.40(2H,m),2.30(1H,m),1.80(1H,m),1.52
(1H,ddd,J=8.0,11.5,23.5Hz),1.31(3H,d,J=7.0Hz),1.10(3H,s)ЁЃдЊЫиЗжЮі(МЦЫужЕ/ЪЕВтжЕ%)ЃК
C 50.59/50.54ЃЌH 6.56/6.52ЁЃ
*ЭЈбЖСЊЯЕШЫ
ВЮПМЮФЯз
1 Hutchinson JH, Money TЃЌPiper SE. Ring cleavage of camphor derivatives:formation of chiral synthon for natural product synthesis.Can J.Chem, 1986, 64ЁУ854
, АйФДвНвЉ
2 Clase JAЃЌMoney T. An enantiospecific route to C, D ring synthon for steroid synthesis. Can J.Chem, 1992, 70ЁУ1537
3 Hutchinson JHЃЌ Money T. Stereoselectivity of C(3)-methylation of camphor and derivatives. Can J. Chem, 1984, 62ЁУ1899
4 Li SWЃЌ Fan RL. Handbook of practical chemistry. Shanghai:Shanghai sciences and technology press. 1981, 549, АйФДвНвЉ(ЙмУЕ ЮтгТ*)